Products
Products
Whole Transcriptome
Evercode WT Mini
Get started with single cell
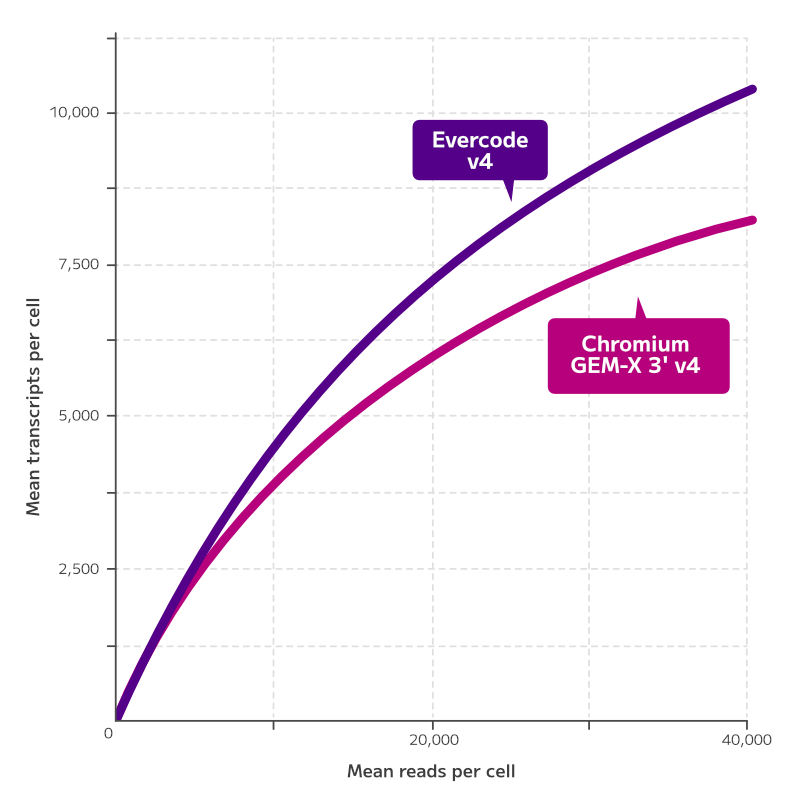
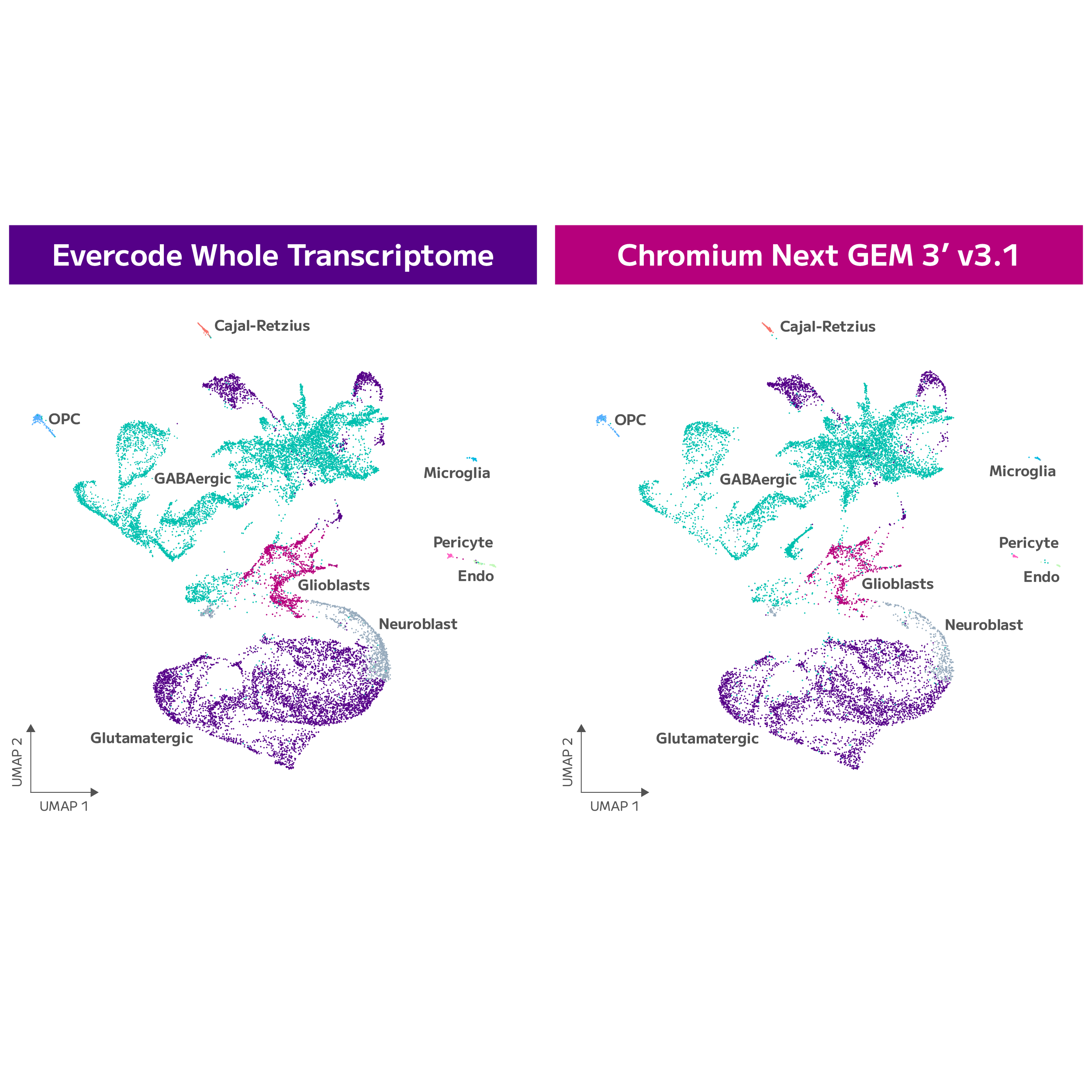
Evercode WT
Achieve your single cell ambitions
Evercode WT Mega
A simpler way to scale your single cell projects
Evercode WT Penta
Scale single cell experiments without compromise
Resources
Resources
Resources
Explore our collection of Evercode resources
Publications
Publications featuring Evercode in single cell research
Product Literature
Delve deeper into the product details
Customer Datasets
Customer datasets covering various applications and sample types
Videos
Watch videos about the Evercode technology
Datasets
Datasets covering various applications and sample types
Experiment Planner
Plan your next single cell experiment
Trailmaker
Streamlined single cell data analysis
Automation
Scalable automation solutions for the Evercode workflow
Support Suite
User guides and additional information for all our products
Company
Company
About Parse
Providing researchers single cell sequencing with unprecedented scale and ease
Upcoming Events
Conferences and in-person meetings
Blog
Customer interviews, tips and tricks, and more
Distributors
Our single cell solutions are available via a growing network of authorized distribution partners.