

Discoveries Hidden in Archived Samples
Major clinical milestones were possible because of archival FFPE samples.
FFPE samples are fixed in formalin and embedded in paraffin, an inexpensive tissue fixation technique that preserves material for years at room temperature.
In 2004, using FFPE samples from 675 blocks, researchers identified 21 genes that quantified the likelihood of distant recurrence in tamoxifen-treated patients with node-negative, estrogen-receptor–positive breast cancer enabling clinical prediction.
In 2010, a study testing EML4-ALK fusion gene via FISH and RT-PCR on non-small-cell lung cancer (NSCLC) tissue identified eligible patients for a trial that showed dramatic response rates and led to the rapid FDA approval of crizotinib, a targeted drug still today used to treat NSCLC.
In 2019, archival FFPE tissues were instrumental in developing and validating gene expression profiles that changed oncology by allowing clinicians to identify which “high clinical risk” women could safely forgo chemotherapy.
FFPE tissue is the most widely used format for long-term clinical tissue preservation, with an estimated 1 billion blocks stored worldwide. Yet unlocking these archives for single cell RNA sequencing (scRNA-seq) has long been a challenge: fixation degrades and fragments RNA, making standard protocols ineffective.
Here we compare the three main approaches: poly(A)-tailing, probe-based hybridization, and random primer-based reverse transcription (Table 1).
How Does FFPE Processing Affect RNA Quality?
Developed to enable pathologists to preserve and analyze tissue specimens, formalin fixation and paraffin embedding (FFPE) have been foundational to pathology for more than a century. Today, they constitute the largest accessible archive of human clinical specimens, representing decades of diagnostic practice and a diversity of diseases.
In FFPE processing, tissues are first fixed with formalin, which cross-links proteins and nucleic acids, preserving morphological structure. After fixation, the water in the tissue is gradually replaced with alcohol and then with a hydrophobic solvent, allowing the sample to be infiltrated with molten paraffin. Once embedded and solidified, the paraffin block provides long-term room temperature preservation and allows the sectioning of thin layers for histological and immunohistochemical analysis.
While FFPE effectively maintains cellular morphology and protein epitopes, the process is detrimental to DNA and even more severely to RNA. It leads to fragmentation, base modifications (such as methylol addition), and cross-linkage between nucleic acids themselves or between nucleic acids and proteins, which hinders extraction and subsequent amplification.
RNA is highly sensitive to these harsh conditions making DNase treatment a requirement to remove DNA contamination, and long-term storage of FFPE blocks causes progressive RNA degradation over months or years. Furthermore, due to fragmentation, mRNA often loses its poly(A) tail, making oligo-dT priming for cDNA synthesis inefficient. New strategies, such as optimized extraction protocols, specialized kits for cross-link reversal, and non-oligo-dT priming methods, have been developed to overcome these obstacles.
What are the Challenges of scRNA-seq in FFPE Samples?
Although invaluable for retrospective molecular studies, these tissues have long been considered incompatible with high-resolution transcriptomic analysis due to extensive RNA degradation, crosslinking, and loss of poly(A) tails that occur during fixation.
Multiple strategies have been developed to enable scRNA-seq of FFPE samples. All strategies start with a de-crosslinking step to reverse the chemical bonds formed during fixation and make RNA molecules accessible. The methods differ in RNA capture strategies.
Each method has pros and cons, and researchers select them based on the scope of their research.
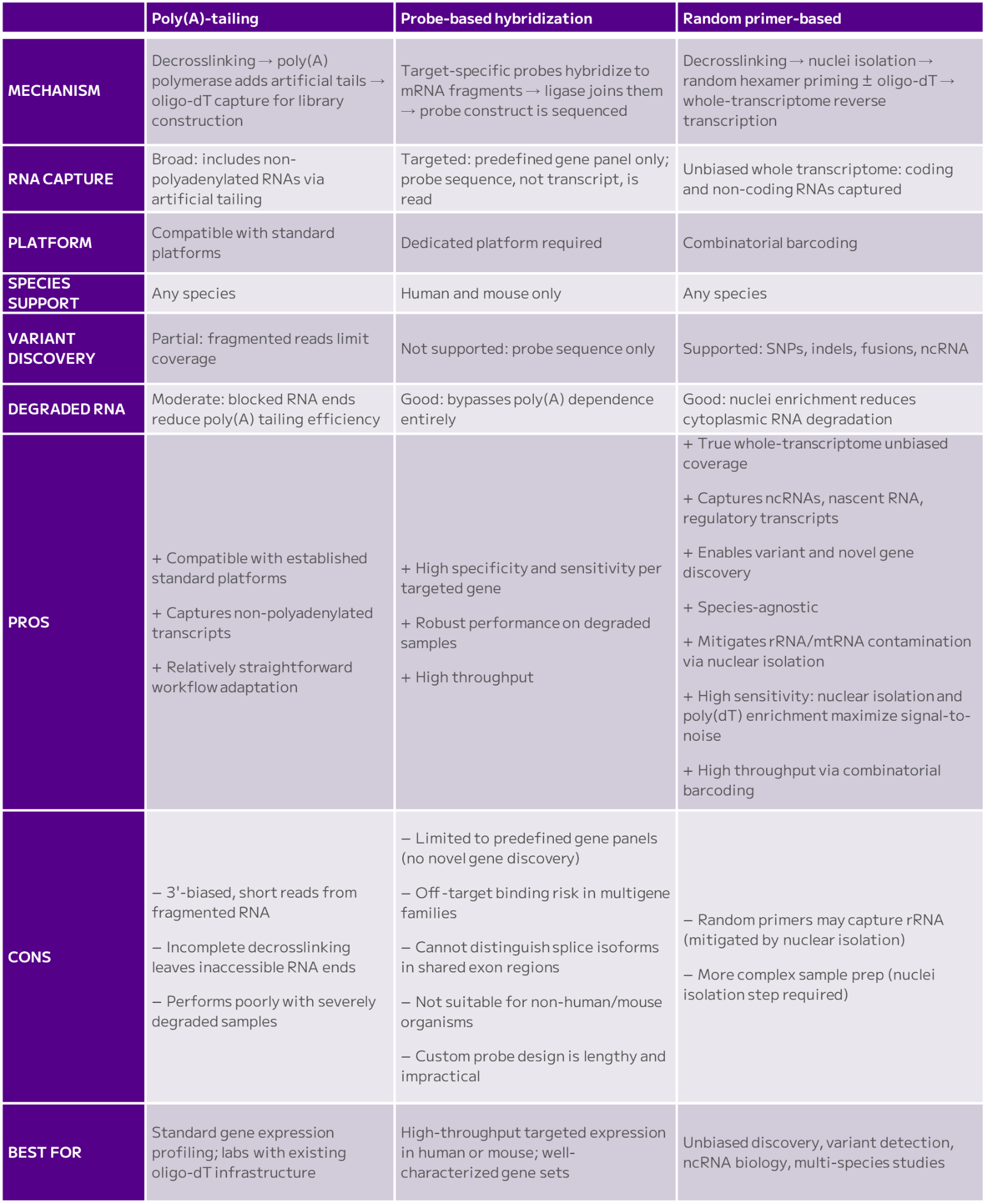
Table 1: The three main approaches for FFPE scRNA-seq, poly(A)-tailing, probe-based hybridization, and random primer-based reverse transcription, have various characteristics, pros and cons. See text.
Poly(A)-Tailing Approaches for FFPE Single Cell Sequencing
Poly(A)-tailing–dependent methods introduce decrosslinking steps to partially restore RNA accessibility, followed by poly(A) polymerase treatment to add poly(A) tails to RNA fragments, enabling subsequent capture via poly(dT) primers used in standard scRNA-seq platforms. Since tails are added artificially, this approach captures a broader range of RNA species, including non-polyadenylated transcripts.
However, because RNA in FFPE samples is heavily fragmented, captured reads represent short, often 3′-biased fragments rather than full transcripts; even when expression levels can be estimated, transcript-level information remains incomplete. Additionally, decrosslinking is rarely complete, leaving some RNA ends chemically blocked and inaccessible to poly(A) polymerase.
Moreover, while these approaches allow the use of established platforms, they struggle with samples exhibiting severe or long-term degradation where the RNA is so fragmented that it may not have enough transcript information.
Probe-Based Hybridization: Benefits and Limitations for FFPE scRNA-seq
Targeted probe-based methods bypass poly(A) dependence by hybridizing matched probe pairs directly to mRNA fragments.
Two target-specific probes are added to the fixed sample and hybridize adjacently to their target transcripts. A ligation enzyme joins them, the resulting ligated construct remains bound to the target RNA and forms the substrate for library construction.
This strategy improves performance on degraded RNA and supports high throughput, but is limited to predefined gene panels and currently restricted to human and mouse transcriptomes. Although customized probe design is possible in principle, the design, development, and validation process is lengthy and laborious, making it an impractical solution for most research applications.
Essentially, the researcher will obtain sequencing reads representing the ligated probe rather than the actual RNA: while the probe accurately reports the gene expression level, the underlying transcript sequence is never read. This makes the approach resembling more an over-expensive microarray rather than RNA sequencing.
This means the method isn’t suitable for any sequence-level biological information that could lead to the discovery of novel genes, splice variants, or unannotated non-coding RNAs.
Limited Species: Like microarrays, probe-based approaches require species-specific probe design. In practice, most commercially available probe panels are optimized for human and mouse samples. This limitation excludes large segments of the biological research community working in veterinary sciences, or in non-mammalian model organisms such as Drosophila melanogaster, Saccharomyces cerevisiae, or Danio rerio.
In these cases, researchers are better off with alternative, more suitable approaches for scRNA-seq of their FFPE samples.
Off Target Binding: The accuracy of gene expression measurements depends on the probes ability to bind its intended target. Indeed, if a probe sequence is also a strong match to another gene, it could lead to an off-target binding. For instance, in gene families with high sequence similarities (like the HLA, or the immunoglobulin, histone genes for example), probes may not be able to distinguish the intended gene and bind to a different one of the same family.
Genes with shared functional domains also may be problematic: if the target is within a conserved functional domain, the probes may bind transcripts from multiple genes that contain that domain.
Alternatively spliced genes share regions. If the probe binds to a shared region, it will not be able to distinguish two isoforms of the same gene.
Losing Cell Identities and Functions: The pre-designed probes are overwhelmingly designed to capture annotated protein-coding exons and a limited subset of well-characterized long non-coding RNAs (lncRNAs). As a result, large fractions of the transcriptome (most non-coding RNAs, untranslated regions (UTRs), intronic sequences, antisense transcripts, and enhancer-associated RNAs) are not captured or sequenced.
Standard expression-focused probe-based panels are fundamentally unsuitable for variant discovery, as sequence information outside probe-targeted regions is permanently lost. For single-nucleotide polymorphisms (SNPs), insertions and deletions (indels), structural variants, and gene fusions only the probe sequence itself is read, not the underlying transcript since the variant is never captured in sequencing reads.
In these cases, variant detection requires purpose-built probe design, as standard expression-focused panels are not designed for this purpose.
Random Primers-Based Methods: Whole Transcriptome Coverage in FFPE scRNA-seq
Some reverse transcription–based approaches adapted for scRNA-seq of FFPE samples employ random primers to capture fragmented RNA without relying solely on intact poly(A) tails or the use of probes.
Random primer-based reverse transcription works particularly well for single nuclei RNA sequencing (snRNA-seq), as it efficiently captures and reverse-transcribes nuclear RNA. Nuclei isolated after decrosslinking are enriched for less-degraded RNA, have reduced cytoplasmic contamination and are less susceptible to dissociation artifacts, improving RNA quality and facilitating transcriptomic profiling of FFPE specimens.
This strategy enables unbiased whole-transcriptome profiling and preserves sequence information across both coding and non-coding RNAs. As a result, it supports unbiased detection of non-polyadenylated RNAs, regulatory transcripts, variants, and novel disease-associated features that probe-based methods can’t capture.
A potential concern with random primers is their lack of specificity, which can lead to ribosomal or mitochondrial RNA detection.
But in the context of snRNA-seq, this concern is substantially mitigated by three factors: nuclear isolation excludes mitochondrial RNA, which is cytoplasmic; the nuclear fraction contains only a small residual amount of ribosomal RNA; and combining random primers with poly(dT) primers enriches for polyadenylated transcripts, further reducing the relative contribution of rRNA.
Together, these features make the combination of nuclei isolation, poly(dT), and random hexamer priming, as implemented in technologies like combinatorial barcoding, an effective strategy for comprehensive transcriptome recovery from FFPE samples.
Why Does Whole Transcriptome Analysis Matter for FFPE Research?
The transcriptome is made of coding and non-coding RNA.
Coding RNA encodes the proteome, but the non-coding RNA plays critical roles in regulation, collectively governing gene expression programs and consequently cell states.
Non-coding sequences serve functional roles and control gene activity. Variants in these regions can be biomarkers for diseases like cancer or neurodegenerative diseases. The majority of variants, from single nucleotide mutations (SNPs) to genomic rearrangements, occur in non-coding portions of the genome. As many of these variants are expressed and therefore detectable at the transcriptomic level, whole-transcriptome RNA-seq becomes a critical discovery tool.
In cancer, for example, long non-coding RNAs have a profound effect on specific cancer hallmarks, and their functions can be altered through genetic and epigenetic changes like SNPs or copy number variants. Since lncRNA can act as oncogenes or tumor suppressors, they correlate with disease predisposition and outcomes (Figures 1a and 1b).
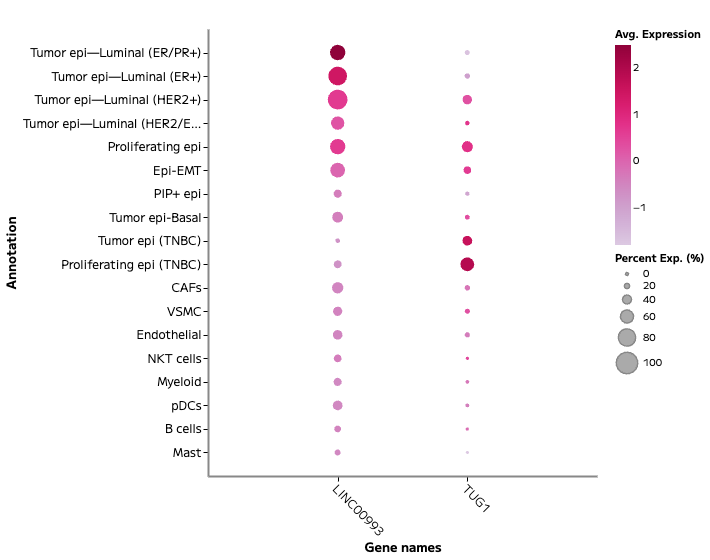
Figure 1a: Whole transcriptome single nuclei RNA sequencing from archival FFPE breast cancer samples resolves stromal and immune compartments, and distinct epithelial programs across clinically relevant subtypes: ER+, ER/PR+, HER2+, and TNBC. This whole transcriptome snRNA-seq experiment identified two lncRNA, LINC00993 and TUG1, associated with breast tumor cell states.
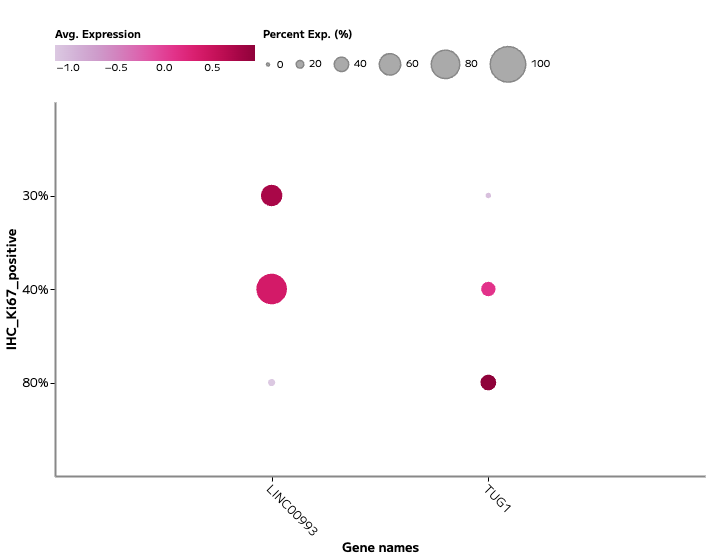
Figure 1b: Expression of lncRNA TUG1 was associated with proliferating epithelial populations (marked by Ki67 expression), linking long non-coding RNA activity to cell cycle state.
An a priori selection of transcripts will exclude analyses of an overwhelming majority of variants occurring in the non-coding portions of the genome, defeating the purpose of a systematic, unbiased interrogation of virtually millions of FFPE samples, and distorting analyses toward already characterized gene-centric effects.
Unlocking the FFPE Archive: The Future of Clinical Transcriptomics
The ability to unlock a much larger resource of archived clinical FFPE samples, once considered incompatible with high-resolution molecular profiling, is transforming decades of preserved specimens into actionable datasets.
As a result, researchers gain unprecedented access to diverse disease stages, rare pathologies, longitudinal cohorts, and real-world clinical heterogeneity, resources that are often impossible to assemble prospectively.
Ultimately, robust scRNA-seq from FFPE samples maximizes the scientific value of existing biobanks and accelerates the development of precision medicine approaches that are grounded in rich, historically inaccessible human tissues.