


What causes cancer? A prevalent narrative is that a mutation in one or more critical genes causes a cell to start growing uncontrollably, eventually forming a tumor. While the mutational changes are undeniably important, it is becoming clear that they are probably not the only driver and something else must be happening in the cell that enables these changes to take hold.
Dr. Katerina Gurova believes that chromatin alterations is an important feature of aggressive cancers.1 In 2009 Dr. Gurova published the first of a series of seminal papers describing a class of newly discovered small molecules, curaxins, able to alter the chromatin remodeling and induce apoptosis in cancer cells.2, 3
Chromatin alteration involvement in cancer is now recognized, and it is believed that it is likely related to changes in the expression of specific genes that drive cancer or stop the control of cell proliferation.
For Dr. Gurova however, these changes may be more general. Besides dysregulated expression of specific genes leading to cancer, she believes that transcription as a whole becomes less controlled. In this less controlled environment, transcription of random genes results in a subset of cells surviving new, changed conditions.
We recently sat down with Dr. Gurova to learn more about the work performed in her lab at Roswell Park Comprehensive Cancer Center, and to discuss how single cell RNA-seq (scRNA-Seq) is helping to shape our understanding of cancer.
Can you tell us about how you came to work on cancer, and more recently, on chromatin?
My overarching goal has been to find anticancer agents since the earliest years. The p53 gene became widely known as a tumor suppressor while I was a graduate student. At the time, it was thought that its discovery would lead to answering our questions about cancer. With this in mind when I first arrived from Russia, I took a position in a lab working with p53. There we developed a system for screening for p53 activators as potential anticancer agents, with a focus on non-DNA damaging compounds.
This led to the discovery of curaxins, small molecules which bind DNA, changing its shape and disrupting chromatin organization.2 Over the years, as I moved into different roles, my quest to understand why these compounds have an anticancer activity has led to my interest in chromatin and what is wrong with chromatin in cancer.
How did problems with the existing paradigm of genetic instability shape your thoughts on chromatin?
When deep sequencing started being widely used, I was shocked to learn that all moles on our skin consist of clones of melanocytes with mutations in BRAF or RAS found in melanoma, but they were not melanoma. These findings suggested that these mutated cell clones were on the way to becoming cancer but we know that it doesn’t happen in the vast majority of cases. Further studies showed similar mutation profiles in other phenotypically normal tissues and organs that did not lead to cancer.
We used mutant HRAS because it is a bona fide transforming agent oncogene involved in many types of cancers and mutant p53 to inactivate endogenous p53.
In your mind, what hypothesis best explains the role of chromatin in oncogene-induced transformation?
I believe tumor cells undergo transcriptional changes much more easily than normal cells because their chromatin is less organized, less constrained. With fewer chromatin constraints, it’s easier for cells to change from one transcriptional program to another.
Normal cells will likely experience such less organized chromatin state when transitioning from one phenotypic state to another – during differentiation, wound healing, inflammation, for example. When transformative events occur during these permissive states, they are more likely to take hold, with random transcriptional changes favoring survival in changing conditions.
How does single cell RNA-seq fit into testing this hypothesis?
Single cell RNA-seq is the only way to distinguish the transcriptional activity of individual cells within a population. Most studies employed bulk RNA-Seq to see average transcriptional activity, but to see and understand our hypothesized permissive state requires looking at individual cells. We hoped that through single cell studies, we would be able to follow the trajectory of cell behavior after the introduction of a transforming agent, gaining insight into the chromatin state.
Could you describe the experimental model you used?
We could not approach this with clinical materials because (1) most samples are already at an advanced stage and (2) it is hard to select good controls as the exact tumor’s cell of origin is often unknown. It can be challenging to separate chromatin instability from genetic instability in such samples, so we needed a straightforward model. One where we could start with normal cells with a shared genome, apply a transforming agent, and observe how the cells responded.
We used HRAS because it is a bona fide transforming agent involved in many types of cancers.4 Importantly, we chose not to apply selection.
When we apply selection, we get transformed clones, but we do not know what happened with the other cells. Did they die because they did not get the transgene? Because they could not withstand the expression of the transgene? Was it something else? Applying selection would leave these cells behind, and we wanted to see what would happen to all the cells – not just those that became fully transformed.
What differences did you see in cell transformation?
Initially, our thinking was that these other cells might die or stop proliferating and that the fully transformed cells would overgrow them. And this could happen eventually. But we were surprised to see that the whole population of cells started to look different, first based on morphology and then by sequencing.
Then, within a couple of weeks, most reverted to look similar, but not identical, to the starting cells. With the sequencing data, we didn’t see a trajectory where cells moved further and further away from the initial state along a linear axis. Instead, we observed more of a circle where cells departed together, with less heterogeneity than expected, then came back to a transcriptome signature closer to where they started. The signature was not the same, but it was similar (Fig 1).
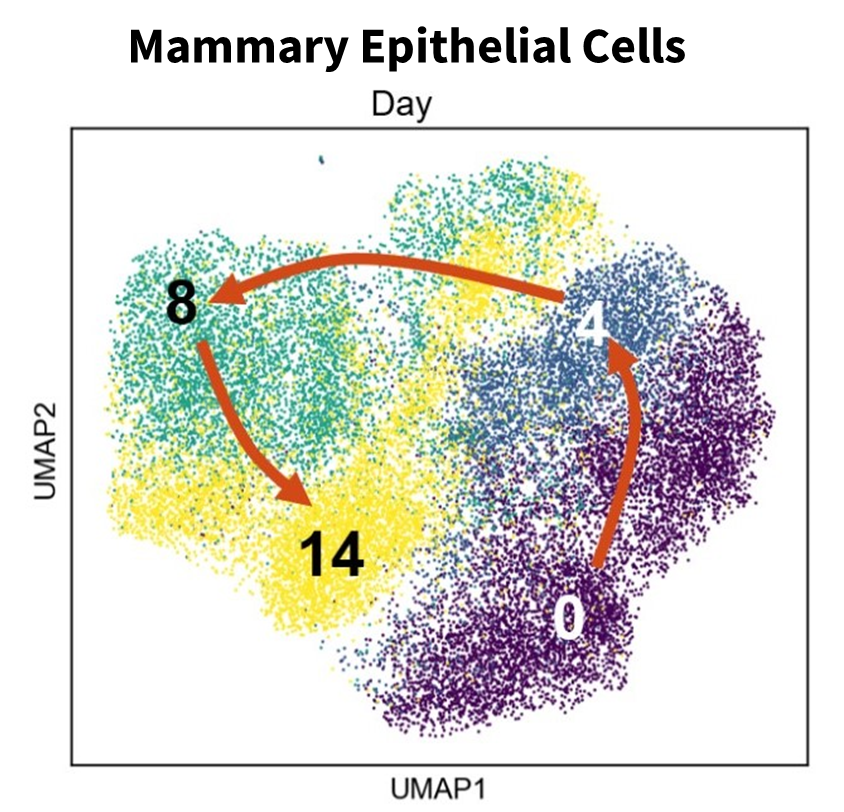
We additionally made several interesting observations. While we expected general gene de-silencing, we were surprised to see a delayed inflammation signature emerging in these cells. We would not have been surprised if this was seen immediately as a result of induction by high levels of HRAS expression, but it appeared to only develop with time in response to secondary events.
We also saw a small number of cells exhibiting activation of interferon that appeared to be transcriptionally stuck. These cells were part of samples at later stages of transformation, but their transcriptome profile was more similar to cells at an earlier stage – suggesting that activation of interferon prevents cells from moving forward to transformation.
Interestingly, in the fibroblasts we studied, we observed a general loss of fibroblast identity and an increase in neuronal markers – despite not exhibiting the typical morphology or traits of neuronal cells. This finding is not completely surprising as the most undifferentiated human cancers often inexplicably express neuronal markers, but why this is the case currently remains a mystery.
What aspects of Parse Biosciences’ Evercode technology did you find most helpful for your research?
One was the ability to fix cells at different time points and then later prepare the libraries and sequence them all together – so we did not have to worry about introducing sample handling differences unrelated to actual biological differences.
Another was that we were looking for technology that did not rely exclusively on prefabricated 3’ primers. While useful when looking for expression of known genes, we were looking for changes in transcription that were not necessarily known or that should not necessarily be there. Parse Biosciences’ inclusion of random oligos made this possible.
A third major consideration was that split-pool combinatorial barcoding reduces the number of doublets and triplets. We expected that some cells in our experiments would have elevated absolute levels of transcription – an overall increase in the number of genes expressed.
If not accounted for, these cells would usually be filtered away in an effort to reduce the likelihood of doublet contamination. But doing so would be removing what we wanted to see. The Evercode technology made the experiment more accurate, increasing the certainty that we were looking at individual cells.
What advice would you give to other researchers interested in using single cell RNA-seq?
Just do it! Single cell RNA-seq is such a powerful technique – it provides so much information and is more accurate.
scRNA-Seq offers a new way of looking at what is happening in biological systems.
You can go from averaging millions of cells to having real granularity and insight into heterogeneity – a crucial factor in understanding the behavior of biological systems.
Looking ahead, what is next?
Besides looking at additional time points for a more granular view of changes along the transformation trajectory, we are currently looking into the transcriptional signature of fully transformed cells.
We wanted an in vitro selection approach that was more controlled than the classical approach of selecting cells able to grow in mice. Two weeks after transformation, we transfer the cells to 3D conditions where they can no longer attach. Both transformed and normal cells can survive this transition, but only fully transformed cells are able to survive subsequent reintroduction to 2D conditions.
We are using single cell RNA-seq to determine the transcriptional profile of these selected, fully transformed cells.
We are also interested in delving into the relationship between inflammation and transformation. Will transformation increase if we induce inflammation? Will it increase if we inactivate interferon? Our studies have provided rich ground for further questions and experiments.
As our understanding grows, we look ahead to a day when we will be able to identify chromatin-related therapeutic approaches that could selectively kill transformation-receptive cells or intervene to prevent chromatin destabilization and avert transformation.
About the Gurova Lab
Dr. Gurova’s lab focuses on identifying novel targets for anticancer treatment and approaches to modulate these targets. Curaxin CBL0137 was among several small molecules with anticancer properties identified by Dr. Gurova’s lab. It is currently being tested in clinical trials.5
For more information, visit https://www.roswellpark.org/research/labs/gurova-lab
Watch this webinar if you want to learn more about Dr. Gurova’s lab and their work.
References:
- Gurova, K. (2019). Chromatin stability as a target for cancer treatment – wiley online library.
- Burkhart C, et al. Curaxin CBL0137 eradicates drug resistant cancer stem cells and potentiates efficacy of Gemcitabine in preclinical models of pancreatic cancer. Oncotarget 5: 11038-11053 (2014). doi: 10.18632/oncotarget.2701
- Gasparian A.V., et al. Curaxins: Anticancer Compounds That Simultaneously Suppress NF-κB and Activate p53 by Targeting FACT. Science Translational Medicine Vol. 3, No. 95 (2011). doi: 10.1126/scitranslmed.3002530
- Schubbert, S., Shannon, K. & Bollag, G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer 7, 295–308 (2007). doi: 10.1038/nrc2109
- CBL0137 for the Treatment of Relapsed or Refractory Solid Tumors, Including CNS Tumors and Lymphoma