

The ability to interrogate the transcriptomic landscape of individual cells has become a reality with the advent of single-cell RNA Sequencing (scRNA-Seq).
The technology has rapidly evolved, expanding our understanding of complex biological processes, development trajectories, and cellular responses.
This series of articles explores the considerations in adopting single-cell transcriptomics for researchers, bioinformaticians, and those vested in the promise of genomics.
Why scRNA-Seq?
The origins of scRNA-Seq, as we know it today, trace back to 2012, with the first generation technology — plate-based methods that unveiled unprecedented sensitivity but offered limited cell throughput.
The technology evolved with second-generation scRNA-Seq methodologies, employing microfluidics and microparticles. This step forward offered increased throughput but required considerable capital investment and is limited to freshly obtained samples that meet the cell-size limitations of the microfluidic device.
The third generation of scRNA-Seq revolutionized the field, introducing combinatorial barcoding.
This strategy uses simple chemistry and the power of combinatorial math, where cells or nuclei undergo multiple rounds of barcoding. The technology bypasses the need for physical cell partitioning and the limiting, expensive equipment.
Combinatorial barcoding introduces a fixation-based approach, which expands its applicability, particularly for long-term studies or clinical samples.
ScRNA-Seq technology allows researchers to examine cell heterogeneity within a tissue to identify rare cell populations. Moreover, with the ability to explore gene co-expression patterns within single cells, scientists can identify co-regulated gene modules and gene-regulatory networks across cells, with implications in all fields of biology and medicine (Fig.1).
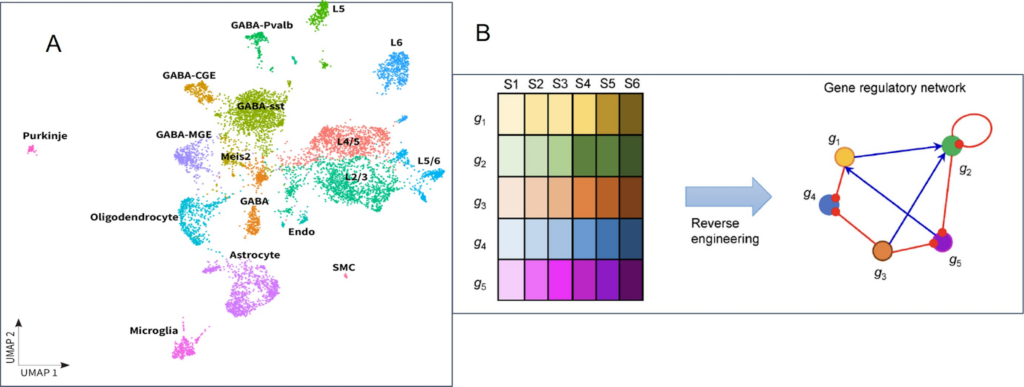
What Insights Does scRNA-Seq Provide?
Unlike bulk RNA sequencing, which enables a look at the averaged gene expression across different cell populations, scRNA-Seq allows a glimpse into individual cell gene expressions (Fig. 2).
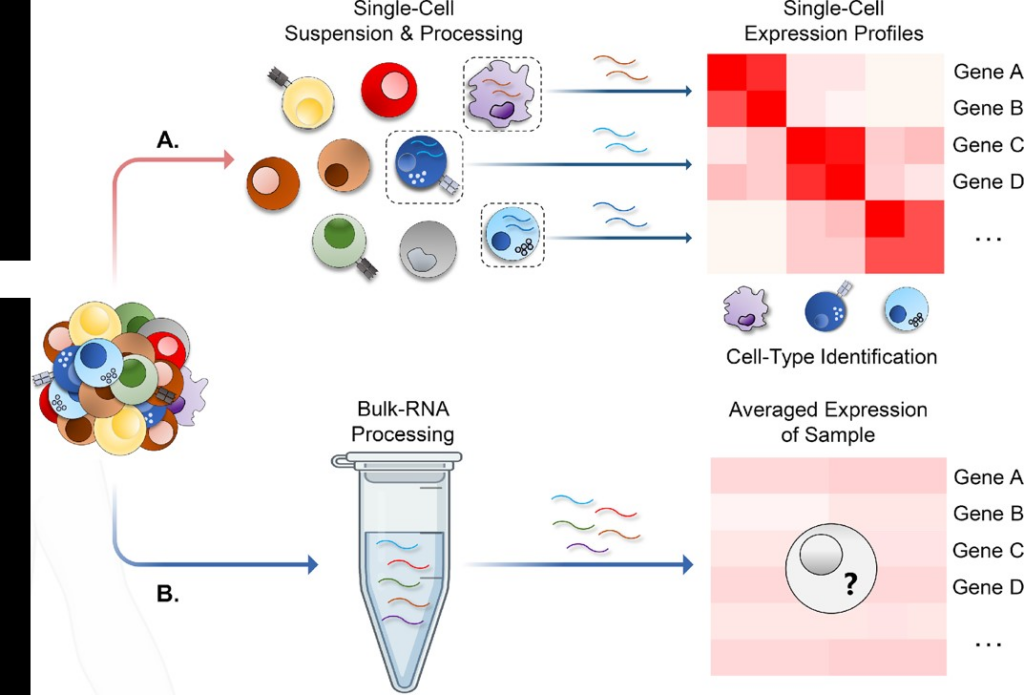
Single cell sequencing helps uncover rare cell types and their roles, leading to insights into gene co-expression patterns vital for understanding cell differentiation and development. This method is instrumental in examining cellular responses to drug treatments and identifying the genes responsible for such reactions.
Researchers have applied this technology in various fields with significant outcomes. For instance, thanks to scRNA-Seq, one significant finding was identifying a rare pulmonary cell type that expresses the transcription factor Forkhead Box I 1 (FOXI1). The absence of FOXI1 in this cell leads to a decrease in Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) expression, which affects mucus physiology, a characteristic feature of cystic fibrosis.
ScRNA-Seq has also been utilized in clinical settings to assess pediatric cancer patients’ reactions to CAR T cell therapies and to understand the therapeutic mechanism of mesenchymal stem cells in treating Crohn’s disease.
Moreover, when using combinatorial barcoding, the incorporated fixation step before processing makes single cell sequencing particularly apt for time-course experiments — due to its scalability and ease of sample storage. Researchers capitalized on the scalability of combinatorial barcoding to conduct an extensive experiment in one single assay, minimizing potential batch variations.
Experimental Design and Sample Preparation in scRNA-Seq
To ensure the success of any project, a clear roadmap and meticulous planning are essential. This is also true for scRNA-Seq experiments.
A successful scRNA-Seq experiment requires clear research questions and adequate resources. The experimental design must cover the sample availability and the number of replicates necessary for journal acceptance.
Sample Selection
The sample availability depends on the type and source of the samples, such as animal, human, and cell lines.
Researchers can utilize fresh or fixed samples, cells, or nuclei. They each come with benefits and challenges.
Fresh samples can present logistical challenges, as they need a lot of planning, resources, and consistency to keep the cells alive and healthy. This may not be sustainable for long-term studies. Conversely, fixed samples are more convenient, as they can be stored for a long time. Moreover, some tissues or organs are unsuitable for single cell analysis — like fresh-frozen samples or highly fibrotic organs. In these cases, it is advisable to pursue nuclei instead.
Another consideration while designing a scRNA-Seq experiment is the sample size, as it affects the statistical power and reproducibility of the results. High-impact journals now require technical and biological replicates as a condition for publication. Pooling may be necessary for small or rare samples.
Sample Preparation Protocol
Optimal cell suspensions require intact cells and RNA, as sample quality affects the efficiency of the critical steps for library preparation — reverse transcription and amplification.
The researcher must utilize the appropriate enzymes, protocols, and techniques for tissue dissociation and cell separation. Each tissue type has different requirements, and it is important to employ a protocol tailored to the tissue type.
The precautions to ensure the highest sample quality don’t have to be complicated. Indeed, simple and practical details that affect the sample quality are temperature, centrifugation speed, aggregation prevention, and management.
Online resources, such as the Worthington Tissue Dissociation Guide and published literature, are valuable resources for optimizing the sample preparation process. Some labs may prefer to invest in commercial instruments like the Miltenyi gentleMACS, which enables the semi-automated dissociation of tissues into single-cell suspensions.
Finally, adding a few QC steps ensures all sample prep steps work as designed. Microscopy and staining techniques are ideal tools to assess cell quality. For example, with a bright field microscope with imaging software, researchers can observe cell morphology and membrane integrity and process and count viable cells from bright-field images.
Data Analysis Approaches in scRNA-Seq
Data analysis is the most exciting part of the experiment. The analytical landscape in scRNA-Seq involves quality control steps and computational methods, from normalization methods and dimensionality reduction to clustering and differential expression analysis.
Demultiplexing is the first step, during which each sample or sublibrary is associated with its respective barcode data.
The experimental QC is the next step. The total cell count shows if the number of cells sequenced aligns closely with the number of intended cell output from the assay. Calculating sequencing saturation helps determine the extent of the library’s complexity sequenced during the experiment.
Moreover, multiplets and cell clumping are sources of errors. Multiplets are cases where multiple cells have the same barcode, which can skew expression data. Cell clumping is a phenomenon where cells stick together and get the same barcode, which can reduce data quality and accuracy.
These issues need to be detected and corrected before proceeding with further analysis.
The next step delves into data analysis. Visualization tools use dimensionality reduction to visualize cell clusters and analyze the gene count matrix to help answer questions like how cells cluster, which cell populations emerge, and which genes have differential expression levels.
More advanced analytical tools, such as Seurat, Scanpy, and scvi-tools perform detailed interpretations, produce high-quality data visualizations, and assign cell types to clusters based on differentially expressed genes. Tutorials on using these tools are readily available on most scRNA-Seq websites.
In the next three articles, we will examine scRNA-Seq experimental design and analysis in detail, providing answers to some scRNA-Seq frequently asked questions.