

For many biologists, happy cells = happy life.
In single cell RNA-sequencing (scRNA-Seq), a high quality cell or nuclei suspension is a primary driver for success.
A high yield of viable cells without debris or clumping enables accurate measurements, high resolution, and coverage — minimizing technical noise. When working on a study, these benefits extend to reproducibility and comparability of the data.
Tissue sources — liquid biopsies, tissue biopsies, organs, cell lines — have different structural characteristics. Producing high-quality single cell suspensions necessitates tailoring the dissociation protocol for each step in the process.
There are some challenges to consider during a cell or nuclei preparation protocol. The seven most important factors for researchers to consider when preparing a single cell suspension for scRNA-Seq (or a single nuclei suspension for snRNA-Seq) are:
- Single cell or single nuclei
- Sample source
- Tissue dissociation
- Temperature
- Cell size
- Cell viability
- Contamination
1 - Single Cell or Single Nuclei
The research question or technical limitations often dictate the decision to analyze cells or nuclei. There are trade-offs in both cases.
Analysis of single cells is appropriate for gaining a comprehensive transcription profile of both nucleus and cytoplasm, to capture cell-cell heterogeneity, identify cell type specific markers, and discover rare cells. It also enables the detection of alternative splicing events or genes highly expressed in the cytoplasm only. Moreover, it allows the examination of molecular pathways, capturing developmental transitional states and dynamics.
But with whole cells, there are a few technical challenges.
Some tissues are fibrous in nature, and harsh dissociation methods cause the loss of delicate cells, alterations in gene expression, and RNA degradation. Single nuclei RNA sequencing may be a safer alternative to single cells in these cases, as the process of generating nuclei suspensions is typically quicker and performed at colder temperatures when compared to the process of dissociating cells.
Sequencing nuclei is a common compromise for researchers using droplet-based microfluidic devices, as these instruments don’t work well with cells larger than 30 µm. The droplet size typically ranges from 30-40 µm, which limits the use of larger cells such as neurons, cardiomyocytes, hepatocytes, and muscle cells. In these cases, sequencing nuclei is an unfortunate sacrifice.
Single nuclei RNA-Seq offers other benefits. SnRNA-Seq enables the capture of greater cellular diversity, including rare cells that may be lost during tissue digestion. Moreover, mitochondrial and ribosomal sequences are more straightforward to remove from single nuclei RNA-Seq data.
2 - Sample Source
Sample source deserves specific consideration in sample preparation. Types of tissue cells, the amount and composition of the extracellular matrix, and the presence of necrotic cells will significantly impact the cell suspension protocol.
Because tissue cells interact via adhesion molecules and junctions, their disruption is necessary during dissociation or digestion. Considering the potential impact of sample preparation techniques on cell viability, RNA integrity, and gene expression, preparation protocols are often specific to the sample source.
Challenges exist in every sample, including some of the most used cell sources in scRNA-Seq experiments, such as:
Cell Lines
Certain cell lines grow adherent to the vessel. Gentle enzymatic treatments with TrypLE™ can effectively dissociate the cells from the culture surface without causing damage.
The density and confluence of cell cultures influence dissociation. High confluence or overcrowded cell cultures increase cell-cell adhesion, necessitating longer or more robust enzymatic reactions.
Some cell lines have resistant cell membranes, requiring prolonged incubation or more potent enzymatic treatments.
iPS Cell Colonies
Induced Pluripotent Stem cells (iPS) form densely packed colonies that clump together. When not adequately dissociated, these colonies form cell aggregates, which hinder efficient preparation.
Like other pluripotent stem cells, iPS cells maintain their pluripotency through interactions mediated by adhesion molecules. Researchers must employ enzymes or reagents specifically designed to target these adhesion molecules for successful dissociation.
Culture conditions and their differentiation states influence the sensitivity of iPS cells to cell preparation protocols. Under conditions that promote stronger cell-cell adhesion, such as when cultured in the presence of ROCK inhibitors, iPS cells may exhibit increased resistance to dissociation.
Brain Cells
Brain cells, particularly neurons, are large cells that exhibit intricate and highly specialized structures. Neurons’ extensive dendritic arborizations and long axons make the tissue particularly hard to handle.
A dense extracellular matrix (ECM) maintains brain tissue integrity by providing structural support. Additionally, cells are tightly connected through various adhesion molecules. While these structures are part of the integrity and function of neural circuits, they can impede digestion.
Neurons’ large size and the sternness of their scaffolding are common technical reasons for choosing to analyze single nuclei instead of whole cells, especially when the technology of choice is droplet-based microfluidics.
Indeed, the whole neuron’s size can be well above the droplet’s. Moreover, cell aggregates or the ECM components can clog the microfluidic channels affecting data quality.
Another challenge for brain cells is the presence of a myelin sheath. These lipid-rich membranes wrap around neurons multiple times, providing insulation and regulating cellular functions. These membranes can resist enzymatic digestion.
These membranes are naturally sticky, leading to obstruction when using droplet-based technologies, as they can adhere to the channels. Indeed, many protocols recommend incorporating a myelin removal step when working with brain cells.
Combinatorial barcoding is an instrument-free technology. Without the limitations of partitioning droplets and channels, there are no neuron size limits or risk of experimental failure due to obstructed channels.
Tumor Cells
While cell adhesion poses challenges in all tissues, it’s particularly problematic in tumor cells. The genetic and molecular alterations in tumor cells contribute to their cohesive and invasive properties. These alterations affect the expression of adhesion molecules and ECM components, making dissociation more difficult.
Tumor cells often exhibit fibrous or calcified regions, necrotic areas, or regions with high cellular density. Additionally, variations in lipid compositions and protein expressions impact membrane rigidity.
These characteristics collectively hinder efficient digestion and can result in incomplete dissociation of tumor cells.
Companies that sell tools and reagents for dissociation often offer tested protocols specifically designed for the challenging task of digesting tumor tissues.
Organoids
Organoids are three-dimensional structures that replicate the organization and functionality of corresponding in vivo tissues. They consist of diverse cell types embedded in an extracellular matrix of proteins and polysaccharides. Various cell-cell junctions interconnect these cells, providing structural support and facilitating communication.
Since organoids resemble organs in their cellular organization and structure, they encounter similar challenges during dissociation. Additionally, organoids consist of different cell types with varying sensitivities to dissociation methods.
Due to their complex nature, dissociation requires careful optimization and balance of enzymatic and mechanical conditions to preserve cell viability and cellular identity.
3 - Tissue Dissociation
Researchers employ enzymatic, mechanical, chemical dissociation, or a combination of these methods to dissociate solid tissues. Minimizing the stress on the cells, quickly dissociating at cold temperatures yields high quality suspensions.
To ensure reliable results, reducing the potential unwanted consequences for the sample is the ultimate goal.
Enzymatic Dissociation
Enzymatic dissociation involves using enzymes to break down specific chemical bonds. Common enzymes are TrypLE, collagenase, dispase, and hyaluronidase.
TrypLE is an alternative to trypsin, ideal for separating adherent cells, like cell lines and cultured cells. Even if it is less toxic than trypsin, protocol optimization will address temperature, concentration, and incubation time to produce high viability suspensions with little stress.
Collagenase suits tissues rich in extracellular matrix and collagen, such as skin, cartilage, or fibrotic tissues. Collagenase breaks down the peptide bonds in the collagen, digesting the extracellular matrix and releasing the cells.
Different tissues may require different types of collagenases, even though these enzymes have similar characteristics. For instance, type I collagenase works well for digesting intestines, mammary glands, and endothelial cells. Type II collagenase is more suitable for breaking down cartilage and isolating osteoblasts.
Dispase is a gentle agent that cleaves fibronectin and type IV collagen, making it ideal for preparing skin cell suspensions. It is also well-suited for detaching cell colonies and dissociating tissue pieces into small clumps.
Hyaluronidase breaks down hyaluronic acid-rich extracellular matrices. This enzyme is often used in combination with collagenase. It is also helpful in hyaluronic acid-rich ECMs, such as brain and tumor samples.
Mechanical Dissociation
Mechanical homogenization methods, such as grinding, blending, sonicating, dounce homogenization, or shaking, are employed for larger tissues. They offer reduced variability compared to manual techniques, such as pipette trituration or tissue scraping.
Some homogenization systems, such as the gentleMACS™ Dissociator or the Singulator 100, are more gentle and consistent than manual methods. Proper calibration and testing for each tissue type are necessary to maintain cell viability and prevent over-homogenization.
Chemical Dissociation
Chemical dissociation involves using agents or detergents like EDTA, EGTA, or Triton 100, which disrupt cell-cell and cell-matrix interactions. While less harsh than enzymatic reactions, these agents work better when combined with mechanical or enzymatic methods.
4 – Temperature
Lower temperatures are beneficial for preserving RNA integrity.
Keeping tissues cold slows down the activity of enzymes that can alter gene expression and induce cell death. However, it also slows down the activity of digestion enzymes that typically function optimally at around 37°C, the human physiological temperature.
Therefore, optimizing experimental conditions involves finding the right balance between temperature and enzymatic activity.
5 – Cell Size
Cell size influences the experimental design and can impact downstream data quality.
Large cells influence the efficiency of reverse transcriptase and amplification steps. Large cells generally contain more RNA, generating more complementary DNA (cDNA). This abundance affects the amplification process. Unequal amplification results in overrepresentation or underrepresentation of cDNA molecules in the sequencing library, biasing results.
An often unmentioned technical limitation of droplet-based microfluidics is their inability to capture larger cells. If a cell is too large for the partitioning droplet, it may not fit inside a single droplet or clog the microfluidic channel, which may result in wetting failures. Cells larger than 40 µm significantly raise the risk of experiment failure. Therefore, the recommended cell size for droplet-based methods is 30 µm or smaller.
Cell size also affects microwell-based assays. Cells greater than 30 µm may negatively influence the microwells’ capture efficiency and cell loading rate. This results in cell loss, lower coverage, and contaminated data.
ScRNA-Seq of large cells like neurons or cardiomyocytes benefits from combinatorial barcoding approaches that aren’t limited by cell size. The technology is chemistry-based, bypassing cell size constraints introduced by instrument-centric techniques.
6 - Cell Viability
Some cell death is unavoidable during the preparation process.
Enzymatic methods are known to be effective but can be harsher on cells compared to mechanical or chemical methods. This phenomenon is especially true for challenging tissues such as brain or solid tumors.
Combining mechanical and enzymatic dissociation techniques helps minimize cell death. The presence of dead cells in the sample can have adverse effects on the results.
Dyes like trypan blue can penetrate the cytoplasm of dead cells and enable viability assessment under the microscope before proceeding with the experiment. Fluorescent dyes like propidium iodide (PI) are more accurate. PI binds the nucleic acid of apoptotic cells and is retained in the nucleus, regardless of a cell’s membrane integrity.
7 - Contamination
Contamination is the presence of unwanted material in the sample. Contamination interferes with the protocol progression and the quality of the data output. Significant culprits for contamination are cell debris and ambient RNA.
Cell Debris
Cell debris results from incomplete digestion of tissue fragments or cell death during sample preparation. It consists of cell or membrane fragments and aggregates. The presence of cell debris has significant implications on the accuracy of results in different ways.
Debris from cell fragments or dead cells may be erroneously counted as live and included in the analysis. This introduces noise in the data and skews data representation accuracy.
Larger clumps of debris can clog the channels in microfluidic systems, resulting in failed runs. In microwell-based scRNA-seq assays, large clumps can prevent the capture of the single cell or the barcoded beads. These problems are easy to bypass using hardware-free, plate-based scRNA-Seq technologies like combinatorial barcoding.
Utilizing filtration through strainers can significantly improve sample quality. The appropriate pore size for filtration depends on the size of the cells.
As a general guideline, many protocols recommend a 40 µm pore size, allowing for the maximum diameter of most cells while retaining clumps and debris. However, narrower pore sizes may be more suited for smaller cells. Larger pore sizes (>70 µm) are appropriate for larger cells, such as neurons or cardiomyocytes.
Ambient RNA
Another source of contamination is ambient RNA. Ambient RNA can originate from cell lysis during sample preparation or sources other than cells of interest.
Droplet-based scRNA-Seq technologies struggle significantly with this problem. Damaged or dead cells leak their RNA into the solution during sample preparation. Leaked RNA will be encapsulated in the droplets with the single cell and barcoded with the cell’s native RNA, contaminating the data.
Moreover, empty droplets can erroneously be called cells if they contain enough ambient RNA. This leads to the inaccurate assignment of transcripts to cells and misclassifying empty droplets as cells (Fig.1).
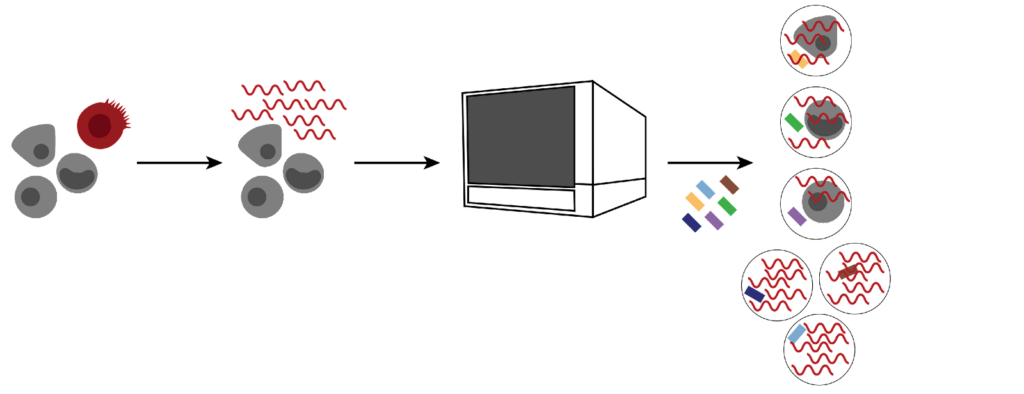
Mitigation strategies to reduce the contamination effects include computational methods to estimate and remove the background noise. Common methods include CellBender, DecontX, and SoupX. These methods are imperfect and have trade-offs that should be well understood before implementation.
The cleanest and most direct approach is combinatorial barcoding, as the method is inherently less susceptible to ambient RNA contamination.
With this approach, the barcoding reaction occurs within the cell/nuclei rather than inside a droplet. For this reason, cells profiled using combinatorial barcoding can undergo a wash step after barcoding has occurred. Therefore, if ambient RNA still makes it through the barcoding steps, it is washed away before downstream library preparation (Fig. 2).
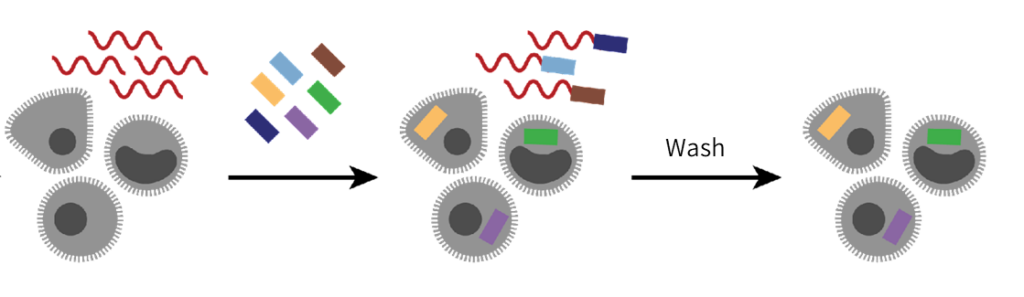
Conclusion
When preparing a sample for a scRNA-Seq experiment, the rule of thumb is “excellence in, excellence out.”
The ideal sample for a scRNA-Seq experiment consists of a homogeneous cell suspension with adequate viability, compatible with the method or platform requirements.
The best quality data provide valuable insights and advancements in cellular research. Taking the time to plan and optimize a sample preparation protocol ensures the validity and reproducibility of experimental results.