Our solution takes you from single cell or single-nuclei suspension through library prep and sequencing and delivers immediate results via our analysis software, Trailmaker.
Our solution takes you from single cell or single-nuclei suspension through library prep and sequencing and delivers immediate results via our analysis software, Trailmaker.
BLOG › Single Cell (r)Evolution › Publication Trends of Single Cell RNA Sequencing Research
Publication Trends of Single Cell RNA Sequencing Research
May 11, 2023
|
7 min read
Updated:May 24, 2024
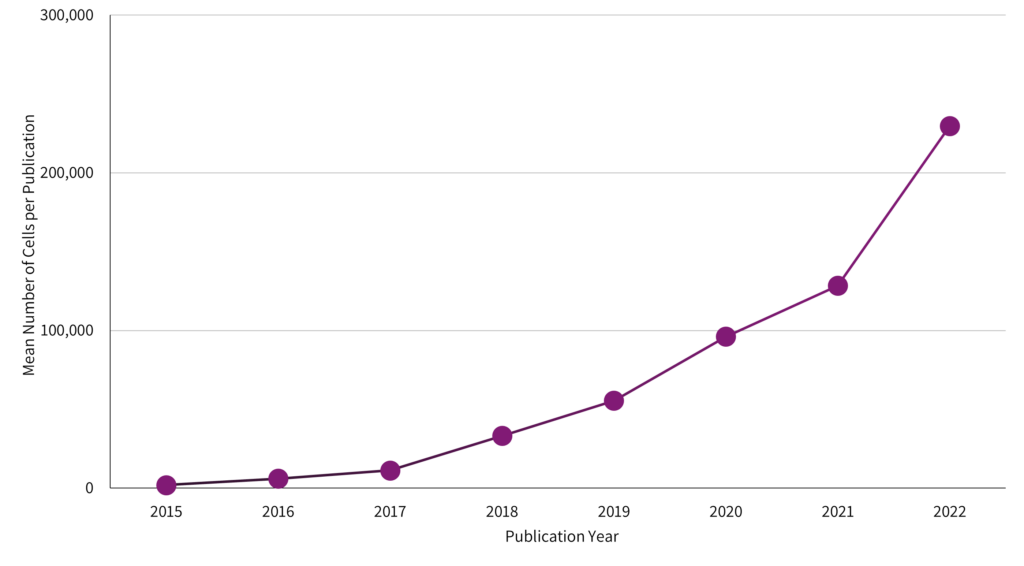
Cells profiled in each publication double every year.
Experimental size is growing at a break-neck pace, demanding increased attention on experimental design and technology selection.
Today’s prevailing methods for single cell RNA sequencing (scRNA-seq) analysis still rely on specialized hardware. Since 2015, these methods have been widely used, allowing tens of thousands of cells to be sequenced in a single experiment. However, limited to a specific instrument, these methods struggle against increasing numbers of samples and cells. Typically, the path to scalability with these technologies is to invest in newer hardware or sacrifice data quality.
The good news is that more efficient and elegant approaches are breaking down these barriers by focusing on chemistry rather than dedicated instruments. Specifically, combinatorial barcoding of single cells supports sample multiplexing and scalability using only essential laboratory equipment, and no commitment to a specific device.
Trends in Publications: Scientists Want More Cells
The need for larger, more detailed studies is reinforced by analyzing current publication trends. The single cell studies database compiled metrics on single cell RNA-Seq publications, allowing for an investigation of how researchers are using the technology. The result of this analysis is stark – the average cell number in publications doubled each year.1
Fig 1: Since 2015, the number of cells analyzed in scRNA-seq publications has grown significantly. Data represent the mean reported cell counts per publication per year. Generated by the author from data in the single cell studies database.
A closer look at these publications reveals that many researchers adapted their research goals into large numbers of small experiments.
A majority of these studies employed droplet-based microfluidic platforms. The approach involves sophisticated hardware to encapsulate individual cells with reagents in droplets. This leads to an inherent inability to scale efficiently, as the hardware limits the number of cells and samples in each run.
For example, cell atlases depend on large cell numbers for comprehensiveness. The Tabula Sapiens Consortium published a molecular reference atlas for more than 400 cell types of the human body. To achieve 500,000 cells from 24 different human tissue samples, the team performed over 150 single cell experiments.2 These results today can be accelerated through significantly fewer experiments, at a fraction of time and resources by using scalable approaches.
To overcome the requirement for multiple costly experiments while ensuring efficient scalability, emerging technologies are now sidestepping the dependency from dedicated hardware. Instead, they enable researchers to scale their experiments effortlessly by utilizing combinatorial barcoding strategies and 96-well plates.
Scaling Enhances Resolution and Statistical Power
Why are scientists chasing higher scale experiments?
Experimental size in scRNA-seq includes two aspects: the number of cells and number of samples. A truly scalable scRNA-seq technology solution accommodates both elements.
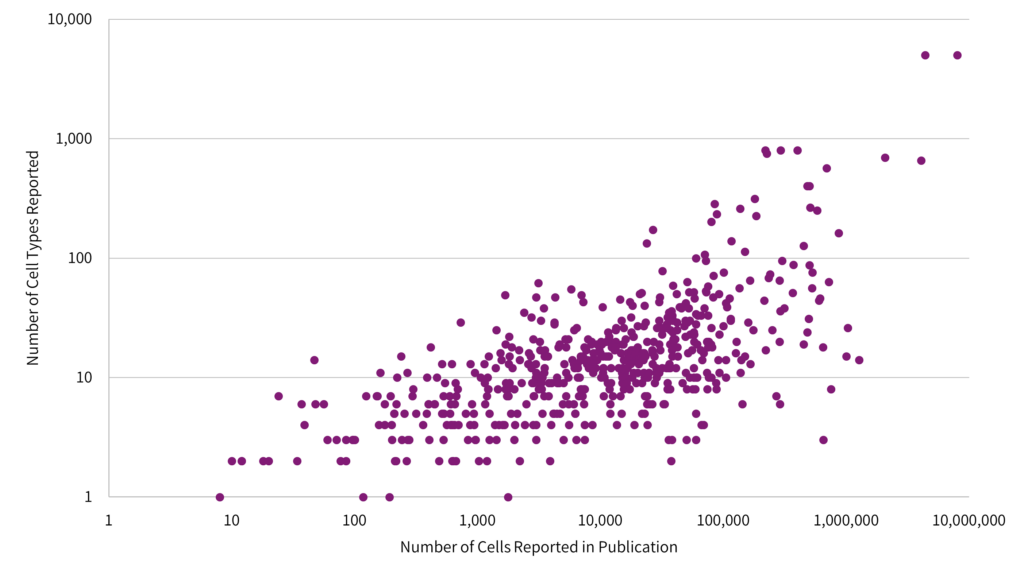
Fig 2: Increasing the number of cells per experiment increases the cell resolution, resulting in a higher number of cells and clusters reported. Data shown in log scale. Adapted from the Single Cell Studies Database.
The data indicate that researchers need to sequence more cells to resolve subtle differences in gene expression profiles across distinct cell types, or to identify lowly expressed cells subsets.
Differential gene expression (DGE) requires large numbers of cells to elucidate distinct cell types. The more cells sequenced, the higher the likelihood to detect subtle differences and capture rare cell subsets. The impact of this effect is especially pronounced in complex tissues.
The capability to capture rare cells is a requirement when studying dormant cancer cells, low frequency lymphocyte clones, rare brain neurons, or transient cell types during cell development. Missing cells due to insufficient number of cells results in biased interpretation of physiological and disease processes.
CRISPR screens have become a powerful source of biological discoveries, with scRNA-seq used to measure the effects of gene edits at the single cell level. A large number of potential gene edits are typically assayed simultaneously with different guide RNAs, requiring numerous cells to acquire statistical significance of each perturbation. Therefore, enough cells must be sequenced to accurately detect changes in gene expression.
Mitigating noise and technical variations during sample processing is a critical aspect of high throughput scRNA-seq that can be addressed by employing more cells. The result is a more accurate representation of gene expression patterns within the studied population.
More Samples Provide More Significance
Robustness is a pillar of unbiased data. Increasing the number of samples in an experiment reduces the impact of technical and biological variability.
Biological replicates are repeated measurements of distinct samples. Their use is an important component of robust studies, as they account for the variability between individuals. By comparing the gene expression from biological replicates, researchers identify differences due to a specific factor — such as disease status, treatment effect, or genetic background.
Early single cell work had few biological replicates as the technology available forced hard choices on experimental complexity. Modern approaches, such as combinatorial barcoding, enable multi-dimensional scRNA-seq experimental designs, allowing for biological variability in populations. With robust experimental designs, biological replicates increase the statistical power of the analysis by reducing false discovery rates and increasing the precision of measurements.
Large sample sizes ensure the accuracy of results and the statistical power to differentiate subtle differences between the gene expression of different cell types. An adequate number of study subjects provides certainty in capturing differences within subject groups and across experimental conditions.
Longitudinal studies present unique challenges as samples are serially acquired over time. Assay reproducibility and batch effects become real concerns with samples prepared on different days, often by different technicians or laboratories. These studies benefit from a scalable method that enables fixation of samples at the time of collection. Samples are then bundled and processed simultaneously to save resources and reduce batch effects.
Moving from Hardware-Based to Chemistry-Based
Since their inception, hardware-based technologies have been the default scRNA-seq method, but they are difficult to scale as sample capacity is tied to the instrument. Practically, this means solution providers force researchers into a persistent purchase, support, and upgrade cycle to meet expanding experimental requirements.
More recent technologies remove the obstacles that limit wider adoption of scRNA-seq by bypassing the requirements of dedicated hardware.
The leading alternative, split-pool combinatorial barcoding, does not require hardware, instead relying on better chemistry. This approach was originally published in Science as SPLiT-Seq, but has been improved significantly and now available commercially as Evercode™.3
The method employs the membrane of the cell or nucleus itself to define the reaction compartment. Combinatorial barcoding bypasses the restrictions of serially encapsulating a cell, bead, and reagents in a droplet, eliminating the scale constraints of a hardware-based system. Unique barcodes are appended to each transcript in a cell so results are traceable back to the origin cell. The entire process uses 96-well plates and standard lab equipment, reducing time and cost barriers to implementation.
Selecting a scalable single cell technology is more important than ever.
Today’s published scRNA-seq experiment requires hundreds of thousands to millions of cells to deliver compelling conclusions. Adopting the right approach for this depth leads to more substantive findings while accelerating research efforts through fewer experiments.
Combinatorial barcoding approaches, such as the Parse Biosciences Evercode technology, accommodate scale more effectively than the hardware constrained technologies that supported the early, foundational efforts.
References
Svensson V, da Veiga Beltrame E, Pachter L. A curated database reveals trends in single-cell transcriptomics. Database (Oxford). 2020;2020:baaa073. doi:10.1093/database/baaa073s
Tabula Sapiens Consortium*, Jones RC, Karkanias J, et al. The Tabula Sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science. 2022;376(6594):eabl4896. doi:10.1126/science.abl4896
Rosenberg AB, Roco CM, Muscat RA, et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science. 2018;360(6385):176-182. doi:10.1126/science.aam8999.
About the Author
Laura Tabellini Pierre
Laura Tabellini Pierre, MSc, is a scientific and technical writer at Parse Biosciences with extensive experience in immunology, encompassing both academic and R&D research.