Our solution takes you from single cell or single-nuclei suspension through library prep and sequencing and delivers immediate results via our analysis software, Trailmaker.
Our solution takes you from single cell or single-nuclei suspension through library prep and sequencing and delivers immediate results via our analysis software, Trailmaker.
BLOG › Customer Profiles › Overcoming Obstacles in Differential RNA Isoform Expression at Single Cell Resolution
Overcoming Obstacles in Differential RNA Isoform Expression at Single Cell Resolution
March 20, 2023
|
13 min read
Updated:May 24, 2024
One common trait among scientists is that if there is no path forward, they will create one. This is what PhD student Elisabeth Rebboah did when she needed to effectively sequence long alternative splicing isoforms across multiple samples and conditions.
Most single cell RNA-Seq (scRNA-seq) approaches rely on short-read sequencing. But when isoforms resulting from alternative splicing hold the key to your research, you need a different approach.
Alternative splicing is a key mechanism by which cells create higher protein diversity and maintain adaptability. It allows a single gene to generate a diversity of RNA transcripts, including or excluding coding sequences, to create a variety of isoforms. The resulting proteins can have different functions or properties.
Single cell RNA sequencing holds the promise to detect alternative splicing within individual cells, especially important as this mechanism is often cell and state specific.
Unlike short-read sequencing approaches that typically measure 50-300 bases per molecule, long-read sequencing technologies can read longer fragments, from 1,000 to 20,000 bases. These methods enable full-length transcript detection and transcript-level analysis of alternative splicing processes. Long-read sequencing protocols have been applied to scRNA-seq, but their scalability may be out of reach for processing multiple samples concurrently.
This challenge inspired Elisabeth Rebboah, a PhD student in the Mortazavi lab at the University of California, Irvine, to develop an efficient method for assessing and mapping alternatively spliced full-length transcripts using scRNA-seq.
With co-author Fairlie Reese, Elisabeth developed LR-Split-seq, a method that combines Parse Biosciences’ Evercode single cell combinatorial barcoding technology with long-read sequencing.1 This technique identifies and quantifies transcript isoform’s expression with single cell resolution.
Elisabeth sat down with us and discussed how they used this approach to analyze changes in cell-type RNA isoforms during cellular differentiation processes.
Before delving into the details of your work, can you touch on how alternative transcript isoform expression regulates processes in human cells?
Alternative splicing affects almost all genes in mammals, including 90%-95% of all human genes. This process vastly expands the proteome and increases the functional diversity of cell types.
A transcript can have multiple alternatively spliced messenger RNAs (mRNAs), generating different protein isoforms with distinct or even antagonistic functions. It could alter protein stability or even its localization. For example, if a transmembrane protein is missing binding domains, the actual function of the protein would fundamentally change.
The misregulation of alternative splicing plays a major implication in cell differentiation, developmental stages, and even diseases.
Can you describe the existing technologies for effective alternative splicing sequencing in a single cell, the limitations, and how you bypassed them?
When sequencing alternative splicing, long-read sequencing is critical to detect starting and ending sites and exon-to-exon junctions.
My lab conducted many single cell and single nucleus RNA sequencing experiments using the Bio-Rad ddSEQ platform. We struggled with obtaining enough single cell barcoded cDNA to input in long-read sequencing platforms like PacBio and Oxford Nanopore.
This is about when the first SPLiT-Seq paper was published in Science.2 We realized it was a valid alternative. Around the same time, other labs introduced long-read protocols that used existing technologies to yield enough cDNA to input into PacBio successfully.
But these alternatives were still using technologies requiring access to microfluidic equipment like the 10X Genomics Chromium. Furthermore, there was a limitation in the number of samples that could be processed on such a platform.
There was still the matter of costs: even with a good amount of reads, these methods still needed to use several PacBio flow cells. And that increases the cost significantly. The same read depth with long-reads is much pricier than with short-reads.
One of the great things about Parse’s Evercode technology was the flexibility to set aside a much smaller sublibrary: I liked the use of the plate to pool cells or nuclei and re-distribute them into a sublibrary.
No matter how small, one sublibrary has a good and even representation of all samples. This distribution enabled us to be flexible. It balances the number of cells and the number of reads. In this paper, we were able to use 1,000 cells enabling us to sequence them less.
Why did you choose a skeletal muscle cell line as your model for a long-read sequencing method?
Our lab is interested in skeletal muscle because, along with the brain, it is one of the tissues that undergoes the most alternative splicing compared to other tissues.
We began with C2C12, a mouse satellite stem cell-derived cell line. In vivo, satellite cells are muscle stem cells responsible for muscle development and repair. During differentiation, these satellite cells express different levels of specific transcription factors. Transcription factor Pax7 decreases in expression as the cells differentiate, while regulatory factors like Myogenin become upregulated.
Satellite cells will replenish the stem cell pool by having asymmetric division. Some cells will be committed to differentiate fully to myoblasts, while the rest will replenish the satellite cell pool.
The Mortazavi lab has a long history with this cell line. It goes back to Dr. Mortazavi’s days as a grad student at Caltech under Barbara Wold, an expert on the C2C12 cell line. It was reassuring to have her as a collaborator in this paper since she would know if we observed the expected expression changes in the cell line.
These cells undergo evident predictable morphological changes, so we knew if the differentiation worked before running experimental tests.
You adopted the Parse technology to sequence long-reads effectively in this benchmarking work. Can you briefly describe the methods?
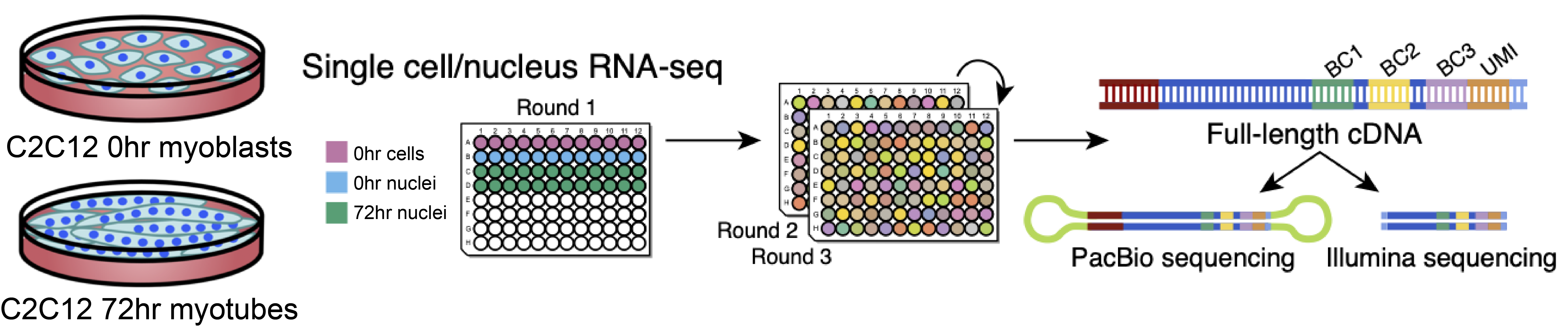
We cultured the C2C12 cell line for 72 hours. Next, we performed single cell/nucleus long-read and short-read sequencing protocols on 0-hour myoblasts and 72-hour differentiating cells. Finally, we prepared the sublibraries using Evercode WT (Fig 1).
Fig 1: Study design: C2C12 cell line was cultured for 72 hours. Cell and nuclei went through the Parse split & pool protocol and analyzed for short-read and long-reads. Image courtesy of E. Rebboah.
We sequenced six, 9,000-cell sublibraries for short-reads. A seventh sublibrary containing a 1,000 cell was split in two: part of the barcoded cDNA was used for the Illumina library preparation, the remainder went through the PacBio long-reads sequencing protocol.
How did you know that the short-read and the long-read sequencing methods had comparable performance?
With the long-read sequencing protocol, we detected the same cell clusters we would see with the short-read protocol. We showed this clustering in three different gene-level UMAPs:
A first UMAP was based on the short-read data,
The second UMAP was based on the long-read data for the same cells,
The third was based on the long-read data on cells annotated by their short-read cluster.
The UMAP structure for all three is similar. I know that some may have issues with UMAPs. But it was convincing that distinct cell types and differentiated cells were grouped in nearly identical clusters between the short and the long-reads, with only minuscule but expected differences between the differentiated cells.
Let’s talk about the results of this experiment: you compare various stages of differentiation in myogenesis and find evidence of gene isoform switching across differentiation stages. Can you elaborate?
The differentiated cells were either Pax7hi – still in a less differentiated, satellite-like state – or Myoghi – on their way to differentiating into muscle fiber. The cells coexisted in both states on the same plate, but there was a clear difference between the cells retaining their satellite cell identity versus the ones that were going to differentiate in the muscle cell.
We did isoform-switching tests across the undifferentiated 0-hour myoblasts and the differentiated 72 h Pax7 and Myogenin high nuclei clusters. We found 21 and 14 significant isoform-switching genes, respectively.
Three examples of isoform switches caught our attention. The Tpm2 locus had increased expression of specific isoforms in the differentiated cells. We also detected Pkm isoforms with mutually exclusive exons corresponding to well-known isozymes.
But Tnnt2 was our favorite because it significantly differed in both transcript length and the transcription start site, the TSS. The differentiated cells mainly used the known TSS corresponding to the longer isoforms.
In comparison, the undifferentiated cells used the shorter isoforms and the corresponding TSS specific for the shorter isoforms. The Tnnt2 gene is a troponin essential for muscle contraction, so it was interesting to see actual biological function.
The Mortazavi lab developed three software tools to analyze long-read sequencing data. How did you use these tools to analyze transcript changes during myogenesis?
We have been doing long-read sequencing for a long time on bulk RNA, and we already had TranscriptClean and TALON before I even got to the lab.
TranscriptClean was written by a previous lab member; it corrects common long-read sequencing artifacts, like indels.3
TALON, or technology-agnostic long-read transcriptome discovery, is for long-read pipelines.4 In this paper, we used it to annotate each read to its isoform of origin and to identify novel transcripts.
And then the last one, Swan, is the newest package that my lab mate Fairlie Reese developed.5 This tool visualizes different isoforms as a genome browser or with a graph-based way of annotating the various splice junctions and the different TSS and TES. Having all the different combinations of the isoforms, you see them in one plot.
Fairlie also implemented differential isoform usage testing in Swan. I highly recommend using Swan for long-read data. It is fantastic. In the paper, Swan was critical in analyzing the relative expression ofTpm2 and Pkm isoforms over the 72-hour period.
Let’s talk about your single cell experience. What aspect of the Parse technology did you find most helpful for your work?
We love Parse so much in this lab. I have a freezer right next to me full of Parse kits.
Many aspects of Parse’s technology are convenient for us – fixing cells or nuclei ahead of time is a game changer for me. Otherwise, especially with tissue samples, it would be a full 24 hours at the bench if we didn’t have this option to fix and biobank ahead of time.
I also mentioned the sublibrary flexibility that enables us to aliquot different numbers of cells or nuclei into different sublibraries. With Parse combinatorial barcoding, one sublibrary has cells from every sample so we can sequence them with long-reads.
Moreover, the plate design flexibility was a game changer. One sample per 96 wells is far from feasible with any other technology, as far as I know. Of course, with 96 samples, there is a trade-off, obviously: fewer cells per sample, but it is still enough. We perform multiple mouse replicates, and with Evercode, we have one replicate per well, which adds power to the data.
Another aspect we love is that there is no need for a microfluidics instrument. It is all multichannel.
And one last thing: having leftovers is highly convenient. We always have plenty of leftover fixed nuclei in our -80C. We can always go back to the same pool of fixed nuclei we sequenced if we need to sequence them again.
We also store barcoded nuclei: we count the nuclei and aliquot them into sublibraries. Most of the time I have barcoded nuclei that I set aside, and when we went back to sequence them, they performed as expected.
We squeeze that kit for all its worth. We love those kits.
What advice would you give to other researchers pursuing gene regulation studies in single cells?
This is a great question. Researchers should have solid, well-framed biological questions before starting a single cell experiment. Unless the goal is more like our goals – more biotech than biology, as we were developing an assay. Or, if the goal is developing a software package that needs single cell data, a simple design may be enough.
Making use of existing data would be great. Some researchers perform single cell experiments, publish the work, and it ends there. Researchers should make more use of the tissue atlases that do exist already. I know it is hard to find the right one sometimes, so I think everyone should make their data publicly available and easily accessible.
For example, our paper’s data is on the ENCODE portal, well organized, and broken out into samples. All our data are there, complete with the experimental details and cell type annotations. It makes it easy for others to access the data and use them.
Another suggestion comes from the wet lab side. Make sure your cell or nuclei preps are of excellent quality. I know from experience that the data will suffer if there is too much debris or cell clumping.
So, take the time to optimize cell preps for different tissues. Before you even start, know what you’re dealing with since different tissues require different preps and techniques.
Looking toward the future, what are you working on now, and what will your next single cell experiment be?
We have a couple of projects.
We are working on a paper highlighting and summarizing all data generated with combinatorial barcoding and microfluidic methods uploaded for the ENCODE Project over the last year.
We have a postnatal time course in mice across five different tissues (two brain tissues, and three others): in this paper, we are focusing only on transcription factors, chromatin regulators, and histone modifiers. We are delving into their role in driving changes in cell types and cell states.
It is a seven-time point, postnatal time course. It entails a heavy analysis of each tissue, a comparative analysis between all the different tissues, and diving deep into regulatory genes. We are trying to tailor this towards ENCODE, which is very transcription factor focused.
Now that we completed our engagement with the ENCODE consortium, our lab is involved with another big consortium, IGVF or Impact of Genomic Variation on Function. We are the mouse group, and we are thrilled to be producing a lot of single nuclei RNA-seq data using Evercode, with the overall goal of identifying cell type-specific expression quantitative trait loci – eQTLs – in mice. So we have an incredible amount of samples coming up. I am thrilled!
The Mortazavi Lab: from the left, Dr. Ali Mortazavi; in the back: Magdalena Gantuz, Elisabeth Rebboah, Fairlie Reese; in the front: Narges Rezaie, Heidi Liang, Jasmine Sakr.
Thank you Elisabeth for speaking with us! Please watch Elisabeth’s webinar to learn more about Parse Biosciences Evercode combinatorial barcoding technology and to gain an in-depth look at the groundbreaking research happening in the Mortazavi lab.
References:
Rebboah, E., Reese, F., Williams K. et al. Mapping and modeling the genomic basis of differential RNA isoform expression at single-cell resolution with LR-Split-seq. Genome Biol (2021);22(1):286. doi: 10.1186/s13059-021-02505-w
Rosenberg, AB., Roco, CM., Muscat, RA., et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science360 (2018). doi: 10.1126/science.aam8999
Wyman, D., Mortazavi A. TranscriptClean: variant-aware correction of indels, mismatches and splice junctions in long-read transcripts. Bioinformatic (2019), 3(2), doi: 10.1093/bioinformatics/bty483
Wyman, D., Balderrama-Gutierrez, G., Reese, F. et al. A technology-agnostic long-read analysis pipeline for transcriptome discovery and quantification. BioRxv (2020) doi: 10.1101/672931
Reese F., Mortazavi A. Swan: a library for the analysis and visualization of long-read transcriptomes. Bioinformatics (2021) 37(9) doi: 10.1093/bioinformatics/btaa836