In the data processing page of Trailmaker Insights module, automatic filtering thresholds are initially set for all datasets. These automatic thresholds can be manually adjusted to tailor them to your experimental design and preferences.
Here are some examples when manual adjustments to filtering should be considered.
Filtering Based on Mitochondrial Content: Considerations for Nuclei Data and Heart Tissue
Filter 3 within the Data Processing page of Trailmaker Insights module removes dead cells based on the proportion of mitochondrial transcripts. A high-quality nuclei sample should have little to no mitochondrial transcripts, but incomplete cytoplasm removal may lead to mitochondrial transcript presence. When working with cells, tissues with high energy demands such as heart tissue, will have a higher number of mitochondria than other cells due to increased cellular respiration. In both cases the default threshold of 3 median absolute deviations may not be the most appropriate filtering strategy.
Filtering Based on Biology: Considerations for Outlier Populations
Filter 4 within the Data Processing page of Trailmaker Insights module removes outliers based on the correlation between number of genes and transcripts. Plotting the number of transcripts versus the number of genes on a logarithmic scale can reveal different cell populations. If a cell population is separated from the main population and has lower genes/transcript expression it is important to understand what these outliers are and decide if they should be eliminated or retained in downstream analysis.
Figure 2 shows a clear population of outliers in a PBMC dataset. They are red blood cells, therefore they can be removed. But they could be a legitimate population like neutrophils, in which case they should be retained for further analysis.
A good strategy for exploring any outlier populations is to disable the filter, reprocess the data and observe where these cells cluster.
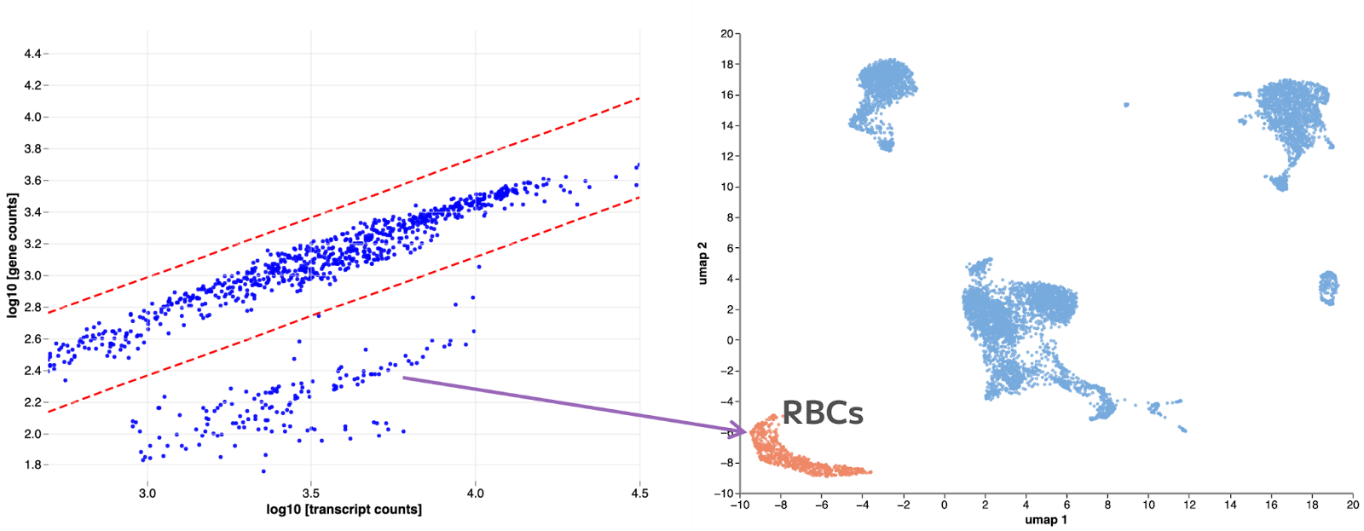
Figure 2: In this example, the population of cells with lower genes and transcripts turns out to be red blood cells, which you might choose to exclude from the downstream analysis.
Filtering Doublets: Considerations for Samples with Low Numbers of Cells
Filter 5 within the Data Processing page of Trailmaker Insights module removes doublets and multiplets from the dataset by simulating artificial doublet profiles using cells from different clusters. It then scores the expression profiles of barcodes in the dataset against the simulated doublet profiles. An important factor to consider in the doublet score calculation is the number of cells in each sample, which dictates the power of the calculation. The reliability of doublet scoring improves with a large number of cells per sample, as the algorithm has sufficient power to better distinguish between singlets and doublets.
When the number of cells per sample is low (e.g., <500), the distribution of doublet scores becomes noisy and more dispersed. In this case, it may not be appropriate to use the default automatic filter setting, and you might consider setting a more appropriate filtering threshold manually, for example by comparing the thresholds across samples where you would expect the doublet rate to be similar.