Trailmaker consists of two major modules that are designed to guide users through the single cell data analysis workflow.
The Pipeline Module is dedicated to processing FASTQ files that are generated using Parse’s Evercode Whole Transcriptome technology.
The Insights Module then performs the downstream analysis and visualization of count matrices. As this module is technology-agnostic it allows multiple data import options (Figure 1).
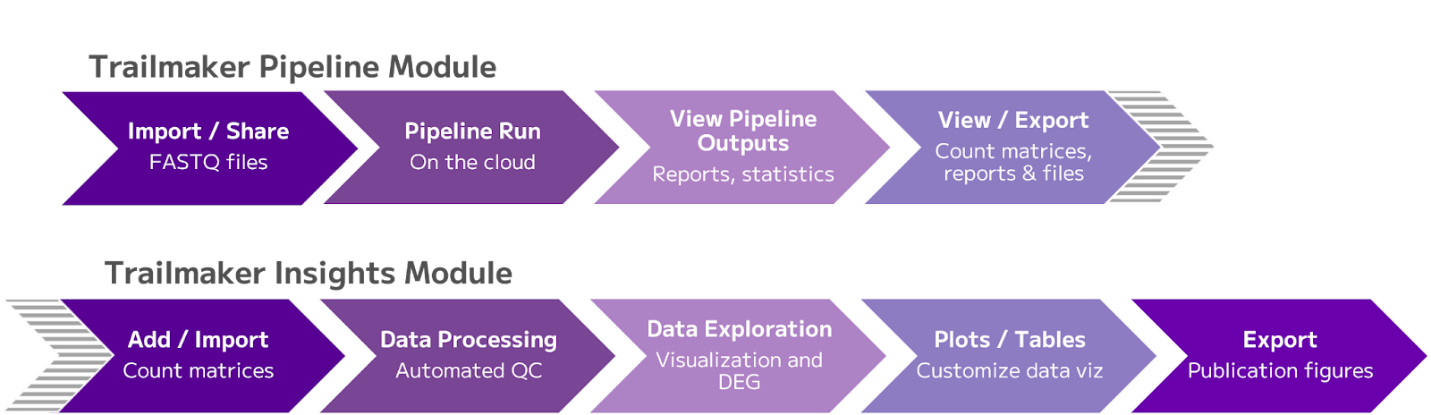
Figure 1: End-to-end data processing in Trailmaker, from FASTQ files to downstream data analysis of gene count matrices
Pipeline Module
Set up a new pipeline run in Trailmaker
The user begins by creating a new run, which opens a step-by-step wizard to guide through the setup process, including input of experimental design details such as Evercode kit type, chemistry version, and the number of sublibraries.
Next, the user uploads the sample loading table—an Excel file that contains the sample loading layout in the first barcoding plate, followed by the selection of the appropriate reference genome, and lastly upload of the FASTQ files.
The runtime of the pipeline run will depend on the project complexity. Therefore, the number of cells and sequencing depth will impact the timing, which can take a few hours when processing a few tens of thousands of cells, to up to 24+ hours when processing millions of cells.
Post-Run outputs and data downloads
Upon completion, the pipeline provides HTML summary reports including statistics like estimated cell counts, median transcript and genes per cell and sequencing quality metrics.
The downloadable data include unfiltered and filtered gene count matrices, pipeline log files, HTML summary reports, as well as intermediate files in the pipeline run including BAM files.
After completion of the pipeline, Trailmaker automatically sets up a project in the Insights module using the unfiltered gene count matrices. A “Go to Insights downstream analysis” button sends the user to the Insight Module project for downstream analysis.
Insight Module
The Insights Module includes three parts – Data processing, Data exploration, and Plots and Tables.
Data Processing
Trailmaker automatically applies a seven-step data processing pipeline that performs all the necessary filtering, integration, normalization and dimensionality reduction steps. The methods and parameters can be adjusted to the user’s preferences.
Filters 1-5 include the removal of background in the classifier and cell size distribution filters, the removal of dead cells based on mitochondrial content, the removal of poor quality cells in the genes-versus-transcripts filter, and doublet removal. Each filter offers manual override options to adjust thresholds and enable/disable filters, allowing customization based on sample characteristics and biology.
The last two steps are data integration—using methods like Harmony, Seurat or Fast MNN—and embedding configuration, where users generate UMAP or t-SNE plots and select clustering algorithms and cluster resolution settings. Normalization and dimensionality reduction also take place during these steps.
Data Exploration
The user can then visualize UMAPs, with options to display different samples, metadata, or gene expression profiles.
To annotate the cell types represented by the clusters, users can run an automatic annotation using ScType by selecting tissue and species. If the automatic annotation is incomplete or unavailable, manual annotation using differential expression and marker gene identification tools is supported.
Differential expression analysis allows comparison between samples/ metadata groups within specific cell types, which can provide biological insight into the dataset. The results can be sorted, filtered for significance, and visualized using heatmaps. Significant genes identified through this process can be exported for pathway analysis directly within the platform.
Plot and Tables
The Plots and Tables Module offers a variety of visualization tools for exploring and presenting the data. All plots available in this module can be fully customized using the sidebar menus to adjust the data/metadata/clusters shown, plot axes, scaling, margins, titles, markers and color schemes to tailor figures for publication.
The types of plots available include:
- Frequency plots to display cluster compositions of samples/ metadata groups by proportion or absolute counts.
- Dot plots, heatmaps and violin plots to display gene expression profiles.
- Embeddings (UMAP or t-SNE) colored by categorical variables like samples/ metadata groups or continuous variables like gene expression.
- Volcano plots showing differential expression between clusters or metadata groups.
- Trajectory analysis showing pseudotime.
Once finalized, plots can be downloaded as high-resolution images.