

Evercode WT v2 Compared with Chromium 3’ v3.1 in Human PBMCs
Experimental Design
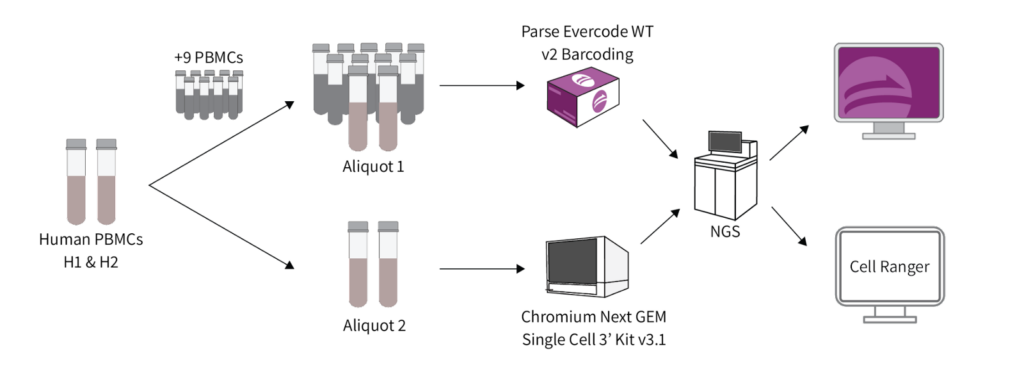
A comparative analysis of scRNA-seq methods with and without sample multiplexing is lacking. Here, we benchmarked methods from two representative platforms: Parse Biosciences (Evercode WT). This method allowed multiple samples in an experiment (multiplexed samples). Chromium 3’ v3.1 from 10X Genomics (Chromium 3’). This method was run with a single sample per lane (no sample multiplexing).
Human PBMCs were used as they are an ideal model system for benchmarking due to their heterogeneity in cell sizes and total RNA content – key factors affecting the performance of scRNA-seq methods. Additionally, the major cell type proportions and their corresponding marker gene expression are well-defined.
PBMCs from two healthy donors (designated as H1 and H2) were prepared in a single batch and distributed into two aliquots. Aliquot 1 was used for 10x library preparation using a Chromium Next GEM Single Cell 3’ Kit v3.1 without multiplexing the H1 and H2 samples. Aliquot 2 was used for Parse library preparation using the Evercode WT v2 kit. Samples H1 and H2 were multiplexed with nine other samples in a single library (total of 11 PBMC samples). All libraries were sequenced together to minimize differences in sequencing depth, with a target of ~10,000 cells per sample.
| Donor | Material | Condition | Number of Cells after QC | Number of Genes (Median) 1 | |
| Evercode | H1 | PBMC | Healthy | 9,819 | 2,319 |
| H2 | PBMC | Healthy | 8,259 | 2,283 | |
| Chromium | H1 | PBMC | Healthy | 6,920 | 1,886 |
| H2 | PBMC | Healthy | 7,020 | 1,983 |
1 Each sample was down-sampled to 20,000 reads per cell. The total number of genes detected was calculated for each cell in the sample, and the median value was taken.
Our study focused on comparing the library efficiency, gene detection sensitivity, gene expression quantification, and cell type recovery between Evercode WT and Chromium 3’ libraries using peripheral blood mononuclear cells (PBMCs).
Key Conclusions:
- Rare cell types (e.g. plasmablasts and dendritic cells) were detected in Evercode WT data only, due to Evercode’s higher sensitivity in gene detection.
- Cell type annotation accuracy is higher in Evercode WT data than Chromium 3’
- Evercode WT showed similar cell type frequencies compared to Chromium 3’ data.
- The addition of multiple samples to the Evercode WT data had no degradation of data quality or usability.
Please refer to our preprint for more details: https://www.biorxiv.org/content/10.1101/2023.06.28.546827v1

"We detected rare cell types with Evercode’s higher sensitivity."
We're your partners in single cell
Reach out for a quote or for help planning your next experiment.