Our solution takes you from single cell or single-nuclei suspension through library prep and sequencing and delivers immediate results via our analysis software, Trailmaker.
Our solution takes you from single cell or single-nuclei suspension through library prep and sequencing and delivers immediate results via our analysis software, Trailmaker.
BLOG › Customer Profiles › Alzheimer’s Disease Research: Using Single Cell RNA-Seq to Better Understand the Brain
Alzheimer’s Disease Research: Using Single Cell RNA-Seq to Better Understand the Brain
February 28, 2023
|
12 min read
Updated:May 24, 2024
June is Alzheimer’s and Brain Awareness Month. It is an opportunity to encourage discussions about dementia and other neurodegenerative diseases.
Alzheimer’s disease (AD), the most common form of dementia, is one of the top 10 leading causes of death according to the World Health Organization. Despite numerous clinical trials, Alzheimer’s disease is the only member of this list with no available drug to treat or slow the disease in any meaningful way.
The advent of high-throughput -omics tools in the last decade has allowed data integration from all biological levels – genes, transcripts, proteins, metabolites. New biological approaches coupled with powerful data analysis methods are uncovering new insights – reviving interest and investments in the disease.
Single cell RNA-Seq (scRNA-Seq) is making new types of experiments possible to better understand this complex disease and how the brain ages normally.
Dr. Vivek Swarup is an Assistant Professor at the Neurobiology and Behavior School of Biological Sciences at the University of California Irvine, where he integrates genomics, transcriptomics, and systems biology approaches to understand the role of transcriptional and regulatory pathways in human neurodegenerative diseases encompassing Alzheimer’s disease (AD).
Recently Dr. Swarup spoke with us about the current research in his lab and his thoughts about the future of AD and single cell research.
What led you to study Alzheimer’s disease using functional genomic approaches?
As a heterogeneous, polygenic disease with hundreds if not thousands of genes involved, Alzheimer’s disease is challenging to model. Unlike disorders typically involving only one or a few genes, AD is associated with multiple genomic mutations, making it challenging and slow to study with traditional approaches.
Functional genomics, along with -omics approaches, makes new hypotheses possible. These can be tested quickly in a high throughput fashion, enabling additional hypotheses and, hopefully, progress.
How does single cell RNA-Seq fit into your work?
The brain comprises multiple cell types: neurons, microglia, astrocytes, oligodendrocytes, etc. Each has its function in the normal brain, but they all change during the course of Alzheimer’s disease.
Single cell analysis lets us understand the independent contribution of different cell types.
Because of the nature of the disease, there are massive changes in the proportion of different cell types throughout its course. Lots of neurons die at the end stage of the disease, with glial cells – like microglia and others – increasing in number.
Bulk gene expression analysis methods will show the effects of these changes, but they will not show what is happening in individual cell types. Single cell RNA-Seq gives us that view.
Your lab developed hd-WGCNA, an R package to enable better insights in high dimensional transcriptomics. What challenges with single cell analysis led to the development of hd-WGCNA?
Understanding gene regulation has been the holy grail of disease research. If we can understand how our 20,000 or so genes function as a group, how they are regulated, and how they change during the course of a disease, we can intervene with drugs and other therapeutic approaches.
WGCNA, which stands for Weighted Gene Co-expression Network Analysis, is a method developed 10 to 15 years ago to identify groups of coregulated genes and their regulators.1 First applied to microarray and then bulk RNA sequencing data, we now wanted to use it with single cell RNA-seq data.
The problem is that scRNA-Seq data are, by the nature of their focus on individual cells, sparse. Sparsity is essentially a big matrix of data where many genes are not shown as expressed, even though they are – their expression is just beyond the current limits of the technology. WGCNA and other approaches have trouble handling this sparsity.
Luckily computational biologists have been dealing with problems of sparse data for years, so we were able to borrow concepts from the field to make WGCNA compatible with single cell data with hd-WGCNA.2
Now, instead of analyzing one cell at a time, we can merge cells with similar transcriptome profiles into metacells – a group of 20 to 50 cells – then perform co-expression network analysis on these small subclusters of cells. This gives us a good idea of what is happening in the different clusters and cell types.
Your research primarily focuses on microglia. What changes do microglia undergo in the early stages of AD?
Understanding microglia’s role is one of the holy grails of any new neurodegenerative disease. Microglia play distinct roles at distinct stages of the disease. In earlier stages, they may be involved in removing plaques and tangles, using their phagocytic activity to clear damaged neurons.
During the progression stage, microglia’s role shifts. TREM2 mediates their anti-inflammatory morphology and profiles when interacting with aggregates of plaques and tangles, but that is only a part of the story. cut We haven’t discovered the exact mechanisms yet.
But we do know that, eventually, microglia start attacking neurons more aggressively. Their morphology changes further. We call them disease-associated microglia, activated stages of microglia, or M1 microglia.
The main point is that change is a gradual transition, not a binary state. They start killing and phagocytizing damaged neurons. The role of microglia depends on the state of the disease we are discussing.
What did you find in your recent study of the human cortex with late-stage Alzheimer’s disease?
As published last year in Nature Genetics, we showed several characteristics that were impossible to detect prior to single cell RNA-Seq.3
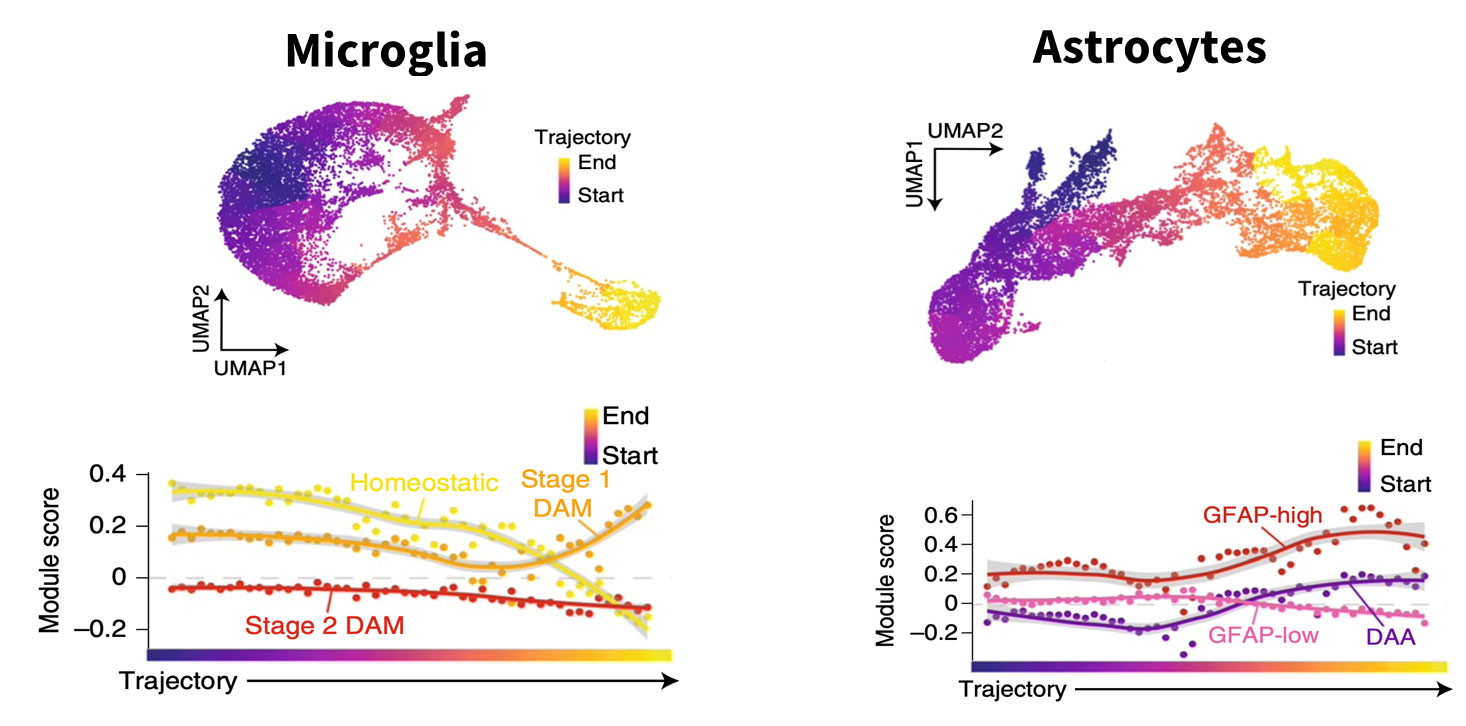
One is the contribution of different cell types to the disease process. Using post-mortem brain samples of individuals clinically diagnosed with Alzheimer’s disease compared to unaffected controls, we observed various changes. And those changes were not limited to neurons, which had been the primary focus of prior research given that they die in large numbers during the course of the disease. All cell types – microglia, astrocytes, oligodendrocytes – were affected (Fig.1).
Fig 1: Single cell RNA-seq analysis allows to “arrange” the cells from controls and disease samples to mimic a time analysis. In this example, by using non-diseased control cells as the starting point, the authors were able to follow the trajectory of Alzheimer’s microglia and astrocyte states. The graphs show genes associated with disease progression changing with time. Figure courtesy of Morabito et al, Nat Genet 2021
We also showed that through pseudotime analysis it is possible to track disease changes in post-mortem samples as if we were tracking them in a living brain.4 To do so, we took all the cells of a single type – microglia from patients who died of Alzheimer’s disease and microglia from unaffected patients – and mixed them together, asking the algorithm to order them according to pseudotime.
Cells from the control samples would all fall at a very early stage with no disease. In contrast, the AD samples, even though they are at a late stage of the disease, would contain a mix of cells – some are fully affected, some are in more intermediary stages, and others are unaffected.
Through pseudotime analysis, we get a picture of the trajectory of change happening across time as if we were looking at the brain of a living individual and tracking the changes happening in each of those cells over the years.
How does what you learned about human microglia in AD fit with data from models of mice that lack microglia?
In the study recently published in Cell Reports, we were interested in understanding the contribution of microglia to Alzheimer’s disease.5 By crossing a strain of mice that genetically lack microglia with a strain of mice that exhibit AD-associated pathologies, our collaborators and we created a mouse model of Alzheimer’s disease where no microglia are present.
To our surprise, we saw a reduction in plaques and tangles but an increase in cerebral amyloid angiopathy (CAA) vascular disease. So, this work shows that microglia are involved in Alzheimer’s disease in mice.
We also know from GWAS experiments that common AD risk variants in the human population are enriched in microglia genes. And we now know that microglia undergo significant changes in the cortex of individuals clinically diagnosed with Alzheimer’s disease.
All this led us to want to better understand the role of microglia in disease progression and whether this could be an avenue for therapeutic intervention.
Did you observe particularly noteworthy changes in cell types other than microglia?
We and others have found considerable changes in oligodendrocytes during Alzheimer’s disease progression. Despite constituting the majority of white matter tracts in the brain, oligodendrocytes are not often studied in detail due to difficulties with cell culturing as well as inherent differences between humans and mice.
We, and others, would like to better understand the role of these changes and how early they happen.
What changes did you observe in astrocytes during Alzheimer’s late stage?
There’s a significant debate about whether or not astrocytes are causal or just contributing to the disease.
Astrocytes, like microglia, gradually change during disease progression. In the late stages, they become defective and do not protect and support neurons as they should. But we still do not know what astrocytes are doing and how they contribute to the disease. Some papers have shown some aspects of astrocytes’ role in AD, but there is much more to learn. Single cell RNA sequencing is at the forefront of understanding their role in Alzheimer’s disease.
What aspect of Parse Biosciences' single cell technology did you find most helpful for your work?
We like to use Parse’s kit for all our single-cell RNA-Seq needs as the protocol is quite simple without using any proprietary instruments. In addition, the cost per sample is also very low compared to other library kits.
Most importantly, the quality of the data generated is quite amazing, with the added advantage that the downstream analysis is also straightforward.
What advice would you give to other researchers pursuing single cell RNA experiments? Are there particular pitfalls to avoid?
Single cell work is exploding so much – everybody is trying to use it to understand their system of interest, which is a good thing.
Parse Biosciences has made it very accessible. Protocols are easy to use, and data analysis is quite easy compared to the past, removing barriers for new users in the community.
While such ease of use is excellent, there is an old adage in the field: “garbage in, garbage out.” Using dead cells or damaged nuclei as input for library creation will compromise the results – but you likely will not even know that something has gone wrong until you have analyzed the data.
It is critically important to use high quality cells or nuclei. I encourage researchers to try different protocols, tuning as needed to the samples being worked with, until they are convinced that they are isolating really good quality cells or nuclei. QC measures that include staining cells with dyes to determine that they are viable or checking for expected nuclei markers can help.
Looking ahead, what are your thoughts on the future of single cell research?
As the technology matures, we will see greater robustness and replicability for each cell, reducing the sparsity we currently try to overcome bioinformatically. Currently, we are seeing 3,000 to 4,000 genes at the median gene expression for each cell, but that should be 10,000 to 12,000 based on what we see in low throughput approaches. We really want to reach the same amount of information with these high throughput methods to get a more complete picture.
Right now, we only see mRNAs and maybe a few non-coding RNAs. Ideally, we will have full coverage of the transcriptome without any biases, like what is currently possible with bulk RNA sequencing. Progress in these two areas will solve numerous challenges.
A third point is that we are now seeing multiomes being generated for single cells with scRNA-Seq, scATAC-Seq, and single cell methylation profiling. Eventually, the field will move forward to understand all these modalities of gene regulation simultaneously.
What are you working on now?
We are trying to build a consensus on changes happening in human and mouse AD using single-cell approaches. What changes occur in oligodendrocytes and in astrocytes? Astrocytes have a great heterogeneity in different brain regions, we want to understand the heterogeneity with respect to the disease.
We are also trying to develop a better mouse model of Alzheimer’s disease. The current most widely used model doesn’t have a neural depth, as it doesn’t have Tau pathology.
It is an amyloid model of Alzheimer’s disease, and we want to develop better mouse models that better reflect the human disease itself.
Can you give us a sneak preview of your next single cell experiment?
We are actually doing library prep today using Parse’s Evercode™ WT v2 kit! It is for samples from the first of the different brain regions, as part of our work to understand what is happening with the different cell types across these different regions.
We thank Dr. Swarup for this enlightening conversation. Watch these two webinars where Dr. Swarup reviewed his research more in depth.
Morabito. S., Reese, F., Rahimzadeh, N., et al. High dimensional co-expression networks enable discovery of transcriptomic drivers in complex biological systems. bioRxiv 2022.09.22 doi:10.1101/2022.09.22.509094
Langfelder, P., Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008). doi: 10.1186/1471-2105-9-559
Morabito, S., Miyoshi, E., Michael, N. et al. Single-nucleus chromatin accessibility and transcriptomic characterization of Alzheimer’s disease. Nat Genet 53, 1143–1155 (2021). doi: 10.1038/s41588-021-00894-z
Trapnell C., et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014 Apr;32(4):381-386. doi: 10.1038/nbt.2859
Shabestari, SK., Morabito S., Danhash EP. et al.Absence of microglia promotes diverse pathologies and early lethality in Alzheimer’s disease mice. Cell Rep 39(11):110961(2022). doi: 10.1016/j.celrep.2022.110961